 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6qu2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of DYRK1A complexed with FC162 inhibitor | ||||||
 Components Components | Dual specificity tyrosine-phosphorylation-regulated kinase 1A | ||||||
 Keywords Keywords | TRANSFERASE / DYRK1A / splicing kinase / kinase inhibitor / Structural Genomics / Structural Genomics Consortium / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of heterochromatin formation / peptidyl-serine autophosphorylation / dual-specificity kinase / [RNA-polymerase]-subunit kinase / tau-protein kinase activity / negative regulation of microtubule polymerization / negative regulation of DNA damage response, signal transduction by p53 class mediator / amyloid-beta formation / negative regulation of mRNA splicing, via spliceosome / G0 and Early G1 ...negative regulation of heterochromatin formation / peptidyl-serine autophosphorylation / dual-specificity kinase / [RNA-polymerase]-subunit kinase / tau-protein kinase activity / negative regulation of microtubule polymerization / negative regulation of DNA damage response, signal transduction by p53 class mediator / amyloid-beta formation / negative regulation of mRNA splicing, via spliceosome / G0 and Early G1 / peptidyl-tyrosine autophosphorylation / cytoskeletal protein binding / RNA polymerase II CTD heptapeptide repeat kinase activity / peptidyl-threonine phosphorylation / protein serine/threonine/tyrosine kinase activity / tubulin binding / positive regulation of RNA splicing / non-membrane spanning protein tyrosine kinase activity / peptidyl-tyrosine phosphorylation / tau protein binding / circadian rhythm / nervous system development / peptidyl-serine phosphorylation / actin binding / protein autophosphorylation / protein tyrosine kinase activity / transcription coactivator activity / histone H3T45 kinase activity / protein kinase activity / nuclear speck / protein phosphorylation / axon / ribonucleoprotein complex / protein serine kinase activity / protein serine/threonine kinase activity / centrosome / dendrite / positive regulation of DNA-templated transcription / nucleoplasm / ATP binding / identical protein binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Chaikuad, A. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Besson, T. / Knapp, S. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: To Be Published Title: Crystal structure of DYRK1A complexed with FC162 inhibitor Authors: Chaikuad, A. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Besson, T. / Knapp, S. / Structural Genomics Consortium (SGC) | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6qu2.cif.gz | 579.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6qu2.ent.gz | 481.2 KB | Display | PDB format |
| PDBx/mmJSON format | 6qu2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6qu2_validation.pdf.gz | 1.4 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6qu2_full_validation.pdf.gz | 1.4 MB | Display | |
| Data in XML | 6qu2_validation.xml.gz | 52.2 KB | Display | |
| Data in CIF | 6qu2_validation.cif.gz | 70.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qu/6qu2ftp://data.pdbj.org/pub/pdb/validation_reports/qu/6qu2 | HTTPS FTP |
-Related structure data
| Related structure data |  4yllS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: _ / Beg auth comp-ID: LYS / Beg label comp-ID: LYS / End auth comp-ID: THR / End label comp-ID: THR / Refine code: _ / Auth seq-ID: 134 - 482 / Label seq-ID: 10 - 358
NCS ensembles :
|
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | Mass: 42081.535 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: DYRK1A, DYRK, MNB, MNBH / Plasmid: pNIC28-Bsa4 / Production host:  |
|---|
-Non-polymers , 6 types, 264 molecules 


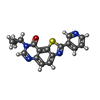


| #2: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-PG4 /  Mass: 194.226 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#4: Chemical | ChemComp-DMS /  Mass: 78.133 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#5: Chemical | ChemComp-JHW /  Mass: 320.368 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H12N4OS Mass: 320.368 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H12N4OS#6: Chemical | ChemComp-EPE / |  Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 241 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.23 Å3/Da / Density % sol: 61.89 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 8.75 Details: 31% PEG 400, 0.25 M lithium sulfate and 0.1 M Tris, pH 8.75 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.2 / Wavelength: 0.9184 Å / Beamline: 14.2 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: Oct 18, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→49.04 Å / Num. obs: 47406 / % possible obs: 98.8 % / Redundancy: 2.9 % / Biso Wilson estimate: 35.9 Å2 / CC1/2: 0.985 / Rmerge(I) obs: 0.124 / Rpim(I) all: 0.098 / Rrim(I) all: 0.173 / Net I/σ(I): 6.9 |
| Reflection shell | Resolution: 2.9→3.06 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.573 / Mean I/σ(I) obs: 1.8 / Num. unique obs: 6853 / CC1/2: 0.661 / Rpim(I) all: 0.448 / Rrim(I) all: 0.794 / % possible all: 99 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4yll Resolution: 2.9→49.04 Å / Cor.coef. Fo:Fc: 0.927 / Cor.coef. Fo:Fc free: 0.889 / SU B: 33.589 / SU ML: 0.3 / SU R Cruickshank DPI: 0.3218 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.368 Details: U VALUES : WITH TLS ADDED HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 117.76 Å2 / Biso mean: 48.558 Å2 / Biso min: 10.12 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.9→49.04 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Refine-ID: X-RAY DIFFRACTION / Type: interatomic distance / Weight position: 0.05
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→2.975 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
