 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3zzm: Crystal structure of Mycobacterium tuberculosis PurH with a novel... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3zzm | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Mycobacterium tuberculosis PurH with a novel bound nucleotide CFAIR, at 2.2 A resolution. | ||||||
 Components Components | BIFUNCTIONAL PURINE BIOSYNTHESIS PROTEIN PURH | ||||||
 Keywords Keywords | TRANSFERASE / HYDROLASE / PURINE BIOSYNTHESIS / TUBERCULOSIS | ||||||
| Function / homology |  Function and homology information Function and homology informationphosphoribosylaminoimidazolecarboxamide formyltransferase / phosphoribosylaminoimidazolecarboxamide formyltransferase activity / IMP cyclohydrolase / IMP cyclohydrolase activity / 'de novo' IMP biosynthetic process / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |   MYCOBACTERIUM TUBERCULOSIS (bacteria) MYCOBACTERIUM TUBERCULOSIS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Le Nours, J. / Bulloch, E.M.M. / Zhang, Z. / Greenwood, D.R. / Middleditch, M.J. / Dickson, J.M.J. / Baker, E.N. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2011 Title: Structural Analyses of a Purine Biosynthetic Enzyme from Mycobacterium Tuberculosis Reveal a Novel Bound Nucleotide. Authors: Le Nours, J. / Bulloch, E.M.M. / Zhang, Z. / Greenwood, D.R. / Middleditch, M.J. / Dickson, J.M.J. / Baker, E.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3zzm.cif.gz | 399 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3zzm.ent.gz | 326.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3zzm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zz/3zzmftp://data.pdbj.org/pub/pdb/validation_reports/zz/3zzm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4a1oC  1pkxS  1zczS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.9711, -0.2038, 0.1243), Vector: |
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 55084.949 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) MYCOBACTERIUM TUBERCULOSIS (bacteria) / Strain: H37RV / Production host: References: UniProt: P67541, UniProt: P9WHM7*PLUS, phosphoribosylaminoimidazolecarboxamide formyltransferase, IMP cyclohydrolase |
|---|
-Non-polymers , 5 types, 409 molecules 

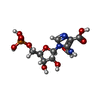


| #2: Chemical | ChemComp-PO4 /  Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 | ||||||
|---|---|---|---|---|---|---|---|
| #3: Chemical |  Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K#4: Chemical | ChemComp-JLN / |  Type: RNA linking / Mass: 367.206 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N3O10P Type: RNA linking / Mass: 367.206 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N3O10P#5: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 404 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46.57 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.5 Details: 24-34% PEG 8000 0.2M SODIUM ACETATE 0.1M SODIUM CACODYLATE PH 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→30 Å / Num. obs: 54907 / % possible obs: 98.1 % / Observed criterion σ(I): 2 / Redundancy: 9.8 % / Biso Wilson estimate: 40.88 Å2 / Rmerge(I) obs: 0.07 / Net I/σ(I): 30.7 |
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 5.8 % / Rmerge(I) obs: 0.4 / Mean I/σ(I) obs: 3.7 / % possible all: 87 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRIES 1PKX AND 1ZCZ Resolution: 2.2→28.76 Å / Cor.coef. Fo:Fc: 0.9438 / Cor.coef. Fo:Fc free: 0.926 / Cross valid method: THROUGHOUT / σ(F): 0 Details: IDEAL-DIST CONTACT TERM CONTACT SETUP. RESIDUE TYPES WITHOUT CCP4 ATOM TYPE IN LIBRARY=K GOL JLN. NUMBER OF ATOMS WITH PROPER CCP4 ATOM TYPE=8147. NUMBER WITH APPROX DEFAULT CCP4 ATOM ...Details: IDEAL-DIST CONTACT TERM CONTACT SETUP. RESIDUE TYPES WITHOUT CCP4 ATOM TYPE IN LIBRARY=K GOL JLN. NUMBER OF ATOMS WITH PROPER CCP4 ATOM TYPE=8147. NUMBER WITH APPROX DEFAULT CCP4 ATOM TYPE=30. NUMBER TREATED BY BAD NON- BONDED CONTACTS=2. RESIDUES A1 TO A3 AND B1 TO B5 WERE NOT MODELLED FOR LACK OF CLEAR ELECTRON DENSITY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 42.57 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.305 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→28.76 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.26 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
