SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED.
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.5418 Å / Relative weight: 1
Reflection
Resolution: 1.45→30 Å / Num. obs: 25810 / % possible obs: 99.7 % / Observed criterion σ(I): 2 / Redundancy: 2.9 % / Rmerge(I) obs: 0.05 / Net I/σ(I): 8.8
Reflection shell
Resolution: 1.45→1.5 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.37 / Mean I/σ(I) obs: 2.5 / % possible all: 100
-
Processing
Software
Name
Version
Classification
REFMAC
5.2.0003
refinement
CrystalClear
datareduction
d*TREK
datascaling
CNS
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDK1PH INSP4 MODEL Resolution: 1.45→25 Å / Cor.coef. Fo:Fc: 0.972 / Cor.coef. Fo:Fc free: 0.956 / SU B: 4.471 / SU ML: 0.073 / Cross valid method: THROUGHOUT / ESU R: 0.089 / ESU R Free: 0.081 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE SIDE CHAINS OF SOME DISORDERED RESIDUES WERE REFINED EITHER WITH THE OCCUPANCY SET TO 0.02, OR THE RESIDUE WAS MUTATED TO ALA. THE ACYL- ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE SIDE CHAINS OF SOME DISORDERED RESIDUES WERE REFINED EITHER WITH THE OCCUPANCY SET TO 0.02, OR THE RESIDUE WAS MUTATED TO ALA. THE ACYL-CHAINS OF THE LIGAND WERE NOT MODELLED DUE TO DISORDER.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.236
545
2.1 %
RANDOM
Rwork
0.182
-
-
-
obs
0.183
25241
99.7 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
















 PDBj
PDBj


















 Assembly
Assembly

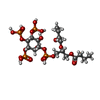
 Mass: 716.350 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H36O22P4
Mass: 716.350 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H36O22P4 Mass: 18.015 Da / Num. of mol.: 163 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 163 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing