 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qgl: Room temperature structure of concanavalin A complexed to bivalen... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qgl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Room temperature structure of concanavalin A complexed to bivalent ligand | ||||||
 Components Components | PROTEIN (SUCCINYLATED CONCANAVALIN A ) | ||||||
 Keywords Keywords | LECTIN (AGGLUTININ) / CONCANAVLIN A / SACCHARIDE BINDING / RECOGNITION COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of defense response to virus / D-mannose binding / defense response / metal ion binding Similarity search - Function | ||||||
| Biological species |   Canavalia ensiformis (jack bean) Canavalia ensiformis (jack bean) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.66 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.66 Å | ||||||
 Authors Authors | Naismith, J.H. | ||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 1999 Title: On the Meaning of Affinity: Cluster Glycoside Effects and Concanavalin A Authors: Dimick, S.M. / Powell, S.C. / Mcmahon, S.A. / Moothoo, D.N. / Naismith, J.H. / Toone, E.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qgl.cif.gz | 107.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qgl.ent.gz | 81 KB | Display | PDB format |
| PDBx/mmJSON format | 1qgl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qg/1qglftp://data.pdbj.org/pub/pdb/validation_reports/qg/1qgl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5cnaS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.68145, -0.00034, 0.73186), Vector: |
-Components
-Protein / Sugars , 2 types, 3 molecules AB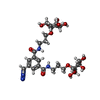
| #1: Protein | Mass: 25622.385 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Details: COMPLEXED WITH BIVALENT LIGAND / Source: (natural) Canavalia ensiformis (jack bean) / References: UniProt: P81461, UniProt: P02866*PLUS#4: Sugar | ChemComp-EJT / |  Type: saccharide / Mass: 659.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H41N5O14 Type: saccharide / Mass: 659.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H41N5O14 |
|---|
-Non-polymers , 4 types, 77 molecules 

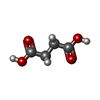

| #2: Chemical |  Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn#3: Chemical |  Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#5: Chemical | ChemComp-SIN / |  Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4 Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 72 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.66 Å3/Da / Density % sol: 66.41 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6 / Details: pH 6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 65 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 293.5 K / pH: 7 / Method: vapor diffusion, sitting drop / Details: Moothoo, D.N., (1998) Acta Crystallogr, D54, 1023. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR591 / Wavelength: 1.5418 |
| Detector | Type: NONIUS / Detector: IMAGE PLATE / Details: MIRROR |
| Radiation | Monochromator: NI / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.66→26 Å / Num. obs: 21164 / % possible obs: 96.8 % / Redundancy: 2.5 % / Biso Wilson estimate: 28.2 Å2 / Rmerge(I) obs: 0.072 / Rsym value: 0.072 |
| Reflection | *PLUS Lowest resolution: 26 Å / Num. obs: 21182 / % possible obs: 96.4 % |
| Reflection shell | *PLUS Highest resolution: 2.66 Å / Lowest resolution: 2.75 Å / % possible obs: 98.3 % / Redundancy: 2.2 % / Rmerge(I) obs: 0.207 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5CNA Resolution: 2.66→26 Å / Rfactor Rfree error: 0.005 / Data cutoff high rms absF: 2919789.43 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: CNS DEFAULT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.66→26 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: RESTRAINTS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.66→2.83 Å / Rfactor Rfree error: 0.015 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 26 Å / % reflection Rfree: 10 % / Rfactor obs: 0.176 / Rfactor Rfree: 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.257 / Rfactor obs: 0.239 |
