 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1pw8: Covalent Acyl Enzyme Complex Of The R61 DD-Peptidase with A Highl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pw8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Covalent Acyl Enzyme Complex Of The R61 DD-Peptidase with A Highly Specific Cephalosporin | ||||||
 Components Components | D-alanyl-D-alanine carboxypeptidase | ||||||
 Keywords Keywords | HYDROLASE / BETA-LACTAM / ANTIBIOTICS / PENICILLIN BINDING PROTEIN / ENZYME / PEPTIDOGLYCAN | ||||||
| Function / homology |  Function and homology information Function and homology informationserine-type D-Ala-D-Ala carboxypeptidase / serine-type D-Ala-D-Ala carboxypeptidase activity / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / proteolysis / extracellular region Similarity search - Function | ||||||
| Biological species |  Streptomyces sp. (bacteria) Streptomyces sp. (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.3 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.3 Å | ||||||
 Authors Authors | Silvaggi, N.R. / Josephine, H.R. / Pratt, R.F. / Kelly, J.A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2005 Title: Crystal structures of complexes between the R61 DD-peptidase and peptidoglycan-mimetic beta-lactams: a non-covalent complex with a "perfect penicillin" Authors: Silvaggi, N.R. / Josephine, H.R. / Kuzin, A.P. / Nagarajan, R. / Pratt, R.F. / Kelly, J.A. #1: Journal: J.Mol.Biol. / Year: 2002Title: Structures of Two Kinetic Intermediates Reveal Species Specificity of Penicillin-Binding Proteins Authors: Mcdonough, M.A. / Anderson, J.W. / Silvaggi, N.R. / Pratt, R.F. / Knox, J.R. / Kelly, J.A. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 2001Title: A 1.2-A Snapshot of the Final Step of Bacterial Cell Wall Biosynthesis Authors: Lee, W. / Mcdonough, M.A. / Kotra, L. / Li, Z.H. / Silvaggi, N.R. / Takeda, Y. / Kelly, J.A. / Mobashery, S. #3: Journal: J.Mol.Biol. / Year: 1995Title: The Refined Crystallographic Structure of a Dd-Peptidase Penicillin-Target Enzyme at 1.6 A Resolution Authors: Kelly, J.A. / Kuzin, A.P. | ||||||
| History |
| ||||||
| Remark 600 | ACYL FORM OF LIGAND H2A The ligand H2A in this structure is in its acylated form. The acylation ...ACYL FORM OF LIGAND H2A The ligand H2A in this structure is in its acylated form. The acylation reaction resulted in the removal of the covalent bond between atoms N5 and C8 of H2A 400, and the formation of the covalent bond between atom OG of SER 62 and atom C8 of H2A 400. MISSING LIGAND H2A ATOMS The following atoms are missing in the coordinate file for ligand CSC 400 in this structure: O1 C1 O2 C26 |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pw8.cif.gz | 168.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pw8.ent.gz | 130.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1pw8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pw/1pw8ftp://data.pdbj.org/pub/pdb/validation_reports/pw/1pw8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1pw1C  1pwcC  1pwdC  1pwgC  3pteS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37422.574 Da / Num. of mol.: 1 / Fragment: DD-PEPTIDASE / Source method: isolated from a natural source / Source: (natural) Streptomyces sp. (bacteria) / Strain: R61References: UniProt: P15555, serine-type D-Ala-D-Ala carboxypeptidase | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-H2A / (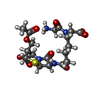  Mass: 487.504 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H27N4O9S Mass: 487.504 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H27N4O9S | ||||
| #3: Chemical |   Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 487 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 487 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.91 Å3/Da / Density % sol: 44.86 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.8 Details: 20% PEG 8000, 50mM Sodium Phosphate, pH 6.80, VAPOR DIFFUSION, HANGING DROP, temperature 298.0K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 1 / Wavelength: 1 Å / Beamline: X12C / Wavelength: 1 / Wavelength: 1 Å |
| Detector | Type: BRANDEIS - B4 / Detector: CCD / Date: Apr 12, 2003 / Details: MIRRORS |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.3→50 Å / Num. all: 82877 / Num. obs: 82877 / % possible obs: 97.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 8.4 % / Rmerge(I) obs: 0.061 / Net I/σ(I): 23.3 |
| Reflection shell | Resolution: 1.3→1.35 Å / Redundancy: 5.1 % / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 3 / % possible all: 81.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 3PTE Resolution: 1.3→10 Å / Num. parameters: 28501 / Num. restraintsaints: 34218 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 0.031
| |||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.06 Å / Num. disordered residues: 14 / Occupancy sum hydrogen: 243 / Occupancy sum non hydrogen: 3067.42 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.3→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
