 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1iki: COMPLEX OF STREPTOMYCES R61 DD-PEPTIDASE WITH THE PRODUCTS OF A S... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1iki | ||||||
|---|---|---|---|---|---|---|---|
| Title | COMPLEX OF STREPTOMYCES R61 DD-PEPTIDASE WITH THE PRODUCTS OF A SPECIFIC PEPTIDOGLYCAN SUBSTRATE FRAGMENT | ||||||
 Components Components | D-ALANYL-D-ALANINE CARBOXYPEPTIDASE | ||||||
 Keywords Keywords | HYDROLASE / PRODUCTS COMPLEX / PEPTIDOGLYCAN / PENICILLIN BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationserine-type D-Ala-D-Ala carboxypeptidase / serine-type D-Ala-D-Ala carboxypeptidase activity / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / proteolysis / extracellular region Similarity search - Function | ||||||
| Biological species |  Streptomyces sp. (bacteria) Streptomyces sp. (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.25 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.25 Å | ||||||
 Authors Authors | Mcdonough, M.A. / Anderson, J.W. / Silvaggi, N.R. / Pratt, R.F. / Knox, J.R. / Kelly, J.A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2002 Title: Structures of two kinetic intermediates reveal species specificity of penicillin-binding proteins. Authors: McDonough, M.A. / Anderson, J.W. / Silvaggi, N.R. / Pratt, R.F. / Knox, J.R. / Kelly, J.A. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 2001Title: A 1.2-A SNAPSHOT OF THE FINAL STEP OF BACTERIAL CELL WALL BIOSYNTHESIS Authors: LEE, W. / MCDONOUGH, M.A. / KOTRA, L. / LI, Z.H. / SILVAGGI, N.R. / TAKEDA, Y. / KELLY, J.A. / MOBASHERY, S. #2: Journal: Biochemistry / Year: 1995Title: BINDING OF CEPHALOTHIN AND CEFOTAXIME TO D-ALA-D-ALA-PEPTIDASE REVEALS A FUNCTIONAL BASIS OF A NATURAL MUTATION IN A LOW-AFFINITY PENICILLIN-BINDING PROTEIN AND IN EXTENDED-SPECTRUM BETA-LACTAMASES Authors: KUZIN, A.P. / LIU, H. / KELLY, J.A. / KNOX, J.R. #3: Journal: J.Mol.Biol. / Year: 1995Title: THE REFINED CRYSTALLOGRAPHIC STRUCTURE OF A DD-PEPTIDASE PENICILLIN-TARGET ENZYME AT 1.6 A RESOLUTION Authors: KELLY, J.A. / KUZIN, A.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1iki.cif.gz | 170.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1iki.ent.gz | 132.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1iki.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ik/1ikiftp://data.pdbj.org/pub/pdb/validation_reports/ik/1iki | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ikgC  3pteS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37422.574 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Streptomyces sp. (bacteria) / Strain: R61References: UniProt: P15555, serine-type D-Ala-D-Ala carboxypeptidase |
|---|---|
| #2: Chemical | ChemComp-REY /   Mass: 303.312 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H21N3O6 Mass: 303.312 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H21N3O6 |
| #3: Chemical | ChemComp-DAL / 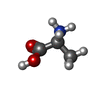  Type: D-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2 Type: D-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 587 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 587 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.97 Å3/Da / Density % sol: 37.6 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.8 Details: PEG 8000, SODIUM PHOSPHATE, pH 6.8, VAPOR DIFFUSION, HANGING DROP, temperature 20K | ||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 1 / Beamline: X12C / Wavelength: 1 |
| Detector | Type: BRANDEIS - B4 / Detector: CCD / Date: Aug 26, 2000 / Details: MONOCHROMATOR, MIRRORS |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.25→20 Å / Num. all: 95204 / Num. obs: 92030 / % possible obs: 96.7 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 5.6 % / Rsym value: 0.069 / Net I/σ(I): 17.2 |
| Reflection shell | Resolution: 1.25→1.29 Å / Redundancy: 2 % / Mean I/σ(I) obs: 2 / Num. unique all: 7323 / Rsym value: 0.178 / % possible all: 77.8 |
| Reflection | *PLUS Lowest resolution: 20 Å / Num. obs: 95204 / % possible obs: 97 % / Num. measured all: 973583 / Rmerge(I) obs: 0.069 |
| Reflection shell | *PLUS % possible obs: 78 % / Rmerge(I) obs: 0.178 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB ENTRY 3PTE Resolution: 1.25→10 Å / Num. parameters: 29573 / Num. restraintsaints: 35362 / Isotropic thermal model: ANISOTROPIC / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: THE ESTIMATED STANDARD DEVIATION OF ATOMIC COORDINATES FOR ALL ATOMS IS 0.045 ANGSTROMS DETERMINED BY INVERSION OF THE LEAST-SQUARES MATRIX.
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-228 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 26 / Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 3215 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.25→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.25→1.29 Å
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 20 Å / Rfactor all: 0.111 / Rfactor Rfree: 0.155 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
