 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1idt: STRUCTURAL STUDIES ON A PRODRUG-ACTIVATING SYSTEM-CB1954 AND FMN-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1idt | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURAL STUDIES ON A PRODRUG-ACTIVATING SYSTEM-CB1954 AND FMN-DEPENDENT NITROREDUCTASE | ||||||
 Components Components | MINOR FMN-DEPENDENT NITROREDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FMN / bioreductive activation / prodrug | ||||||
| Function / homology |  Function and homology information Function and homology information6,7-dihydropteridine reductase / 2,4,6-trinitrotoluene catabolic process / 6,7-dihydropteridine reductase activity / NAD(P)H dehydrogenase (quinone) activity / Oxidoreductases / FMN binding / oxidoreductase activity / protein homodimerization activity / membrane / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Johansson, E. / Parkinson, G.N. / Denny, W.A. / Neidle, S. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2003 Title: Studies on the Nitroreductase Prodrug-Activating System. Crystal Structures of Complexes with the Inhibitor Dicoumarol and Dinitrobenzamide Prodrugs and of the Enzyme Active Form. Authors: Johansson, E. / Parkinson, G.N. / Denny, W.A. / Neidle, S. | ||||||
| History |
| ||||||
| Remark 600 | HETEROGEN FMN 1218 AND CB1 1219 ARE ASSOCIATED WITH CHAIN A. FMN 2218 AND CB1 2219 ARE ASSOCIATED ...HETEROGEN FMN 1218 AND CB1 1219 ARE ASSOCIATED WITH CHAIN A. FMN 2218 AND CB1 2219 ARE ASSOCIATED WITH CHAIN B. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1idt.cif.gz | 102.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1idt.ent.gz | 78.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1idt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/id/1idtftp://data.pdbj.org/pub/pdb/validation_reports/id/1idt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1oo5C  1oo6C  1oonC  1ooqC  1ds7S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23937.182 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | 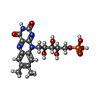  Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P#3: Chemical | 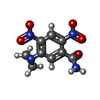  Mass: 252.184 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H8N4O5 Mass: 252.184 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H8N4O5#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 192 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 192 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46.55 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion / pH: 4.6 Details: PEG 4000, sodium acetate , pH 4.6, VAPOR DIFFUSION, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 97 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 0.976262 Å / Beamline: BM14 / Wavelength: 0.976262 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Nov 3, 2000 |
| Radiation | Monochromator: germanium 220 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.976262 Å / Relative weight: 1 |
| Reflection | Resolution: 2→20 Å / Num. all: 31534 / Num. obs: 28476 / % possible obs: 90.34 % / Observed criterion σ(F): 0 / Redundancy: 13.14 % / Rmerge(I) obs: 0.064 / Net I/σ(I): 29.76 |
| Reflection shell | Resolution: 2→2.07 Å / Rmerge(I) obs: 0.201 / % possible all: 98.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1DS7 Resolution: 2→20 Å / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 29.2792 Å2 | ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→20 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| LS refinement shell |
|
