 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1fpy: CRYSTAL STRUCTURE OF GLUTAMINE SYNTHETASE FROM SALMONELLA TYPHIMU... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1fpy | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF GLUTAMINE SYNTHETASE FROM SALMONELLA TYPHIMURIUM WITH INHIBITOR PHOSPHINOTHRICIN | ||||||
 Components Components | GLUTAMINE SYNTHETASE | ||||||
 Keywords Keywords | LIGASE / glutamine synthetase / phosphinothricin / inhibition | ||||||
| Function / homology |  Function and homology information Function and homology informationnitrogen utilization / glutamine synthetase / : / glutamine synthetase activity / protein homooligomerization / manganese ion binding / ATP binding / membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  Salmonella typhimurium (bacteria) Salmonella typhimurium (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.89 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.89 Å | ||||||
 Authors Authors | Gill, H.S. / Eisenberg, D. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2001 Title: The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Authors: Gill, H.S. / Eisenberg, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1fpy.cif.gz | 1.1 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1fpy.ent.gz | 958.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1fpy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fp/1fpyftp://data.pdbj.org/pub/pdb/validation_reports/fp/1fpy | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological complex is a dodecamer. |
-Components
| #1: Protein | Mass: 51744.418 Da / Num. of mol.: 12 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Salmonella typhimurium (bacteria) / Production host: #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: Mn#3: Chemical | ChemComp-ADP /   Mass: 427.201 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#4: Chemical | ChemComp-PPQ / 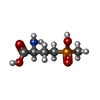  Mass: 181.127 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C5H12NO4P Mass: 181.127 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C5H12NO4P#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1836 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1836 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.35 Å3/Da / Density % sol: 47.71 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: ADP, MPD, spermine, manganese chloride, imidazole buffer, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7 | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 1.1 / Beamline: X12C / Wavelength: 1.1 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Apr 24, 1995 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.89→15 Å / Num. all: 87421 / Num. obs: 87421 / % possible obs: 70 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 14.6 % / Biso Wilson estimate: 53 Å2 / Rmerge(I) obs: 0.074 / Net I/σ(I): 13.5 |
| Reflection shell | Resolution: 2.9→2.95 Å / Redundancy: 1.6 % / Rmerge(I) obs: 0.288 / Num. unique all: 87421 / % possible all: 58.6 |
| Reflection | *PLUS % possible obs: 70 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.89→15 Å / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: The model was built using 12-fold NCS averaged maps and refined using (strict) 12-fold NCS-averaging, or constraints. The phosphinothricin model was restrained to match small-molecule ...Details: The model was built using 12-fold NCS averaged maps and refined using (strict) 12-fold NCS-averaging, or constraints. The phosphinothricin model was restrained to match small-molecule structure solutions (see article). Some of the closely spaced waters are most likely a MPD molecule, such as O61 & O28. A BULK SOLVENT CORRECTION and an anisotropic B-factor correction was applied to the data set (see XPLOR 3.843). Alternate conformations B reflect the movements of catalytic loops upon phosphinothricin binding in the active site, whereas alternate conformations A reflect a native structure. See article for details.
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.89→15 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.843 / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 15 Å / σ(F): 0 / Rfactor obs: 0.248 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
