 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1brr | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | X-RAY STRUCTURE OF THE BACTERIORHODOPSIN TRIMER/LIPID COMPLEX | ||||||||||||
 Components Components | PROTEIN (BACTERIORHODOPSIN) | ||||||||||||
 Keywords Keywords | PROTON TRANSPORT / PROTON PUMP / MEMBRANE PROTEIN / RETINAL PROTEIN / LIPIDS / PHOTORECEPTOR / HALOARCHAEA | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationlight-driven active monoatomic ion transmembrane transporter activity / phototransduction / photoreceptor activity / monoatomic ion channel activity / proton transmembrane transport / plasma membrane Similarity search - Function | ||||||||||||
| Biological species |  Halobacterium salinarum (Halophile) Halobacterium salinarum (Halophile) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||||||||
 Authors Authors | Essen, L.-O. / Siegert, R. / Oesterhelt, D. | ||||||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1998 Title: Lipid patches in membrane protein oligomers: crystal structure of the bacteriorhodopsin-lipid complex Authors: Essen, L. / Siegert, R. / Lehmann, W.D. / Oesterhelt, D. #1: Journal: J.Mol.Biol. / Year: 1993Title: Orthorhombic Crystal Form of Bacteriorhodopsin Nucleated on Benzamidine Diffracting to 3.6 Angstrom Resolution Authors: Schertler, G.F.X. / Bartunik, H.D. / Michel, H. / Oesterhelt, D. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1brr.cif.gz | 151.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1brr.ent.gz | 119.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1brr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/br/1brrftp://data.pdbj.org/pub/pdb/validation_reports/br/1brr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2brdS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS oper:
|
-Components
-Protein , 1 types, 3 molecules ABC
| #1: Protein | Mass: 26740.330 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Details: SCHIFF BASE BETWEEN LYS 216 AND RET 999 / Source: (natural) Halobacterium salinarum (Halophile) / Cellular location: MEMBRANE / Strain: R1 / References: UniProt: P02945 |
|---|
-Sugars , 2 types, 3 molecules 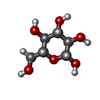
| #2: Polysaccharide | Source method: isolated from a genetically manipulated source #6: Sugar | ChemComp-BGC / |  Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
|---|
-Non-polymers , 4 types, 17 molecules 


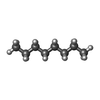
| #3: Chemical |  Mass: 284.436 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H28O Mass: 284.436 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H28O#4: Chemical | ChemComp-ARC /  Mass: 298.547 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C20H42O Mass: 298.547 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C20H42O#5: Chemical |  Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#7: Chemical | ChemComp-OCT / |  Mass: 114.229 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18 Mass: 114.229 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18 |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.18 Å3/Da / Density % sol: 54 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.2 / Details: SEE REFERENCE 2, pH 5.2 | |||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion / PH range low: 5.6 / PH range high: 5.4 | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 1 / Beamline: X11 / Wavelength: 1 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jul 15, 1997 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→25 Å / Num. obs: 18504 / % possible obs: 82.3 % / Observed criterion σ(I): 0 / Redundancy: 2.7 % / Rmerge(I) obs: 0.078 / Net I/σ(I): 10.2 |
| Reflection shell | Resolution: 2.9→3.03 Å / Rmerge(I) obs: 0.215 / Mean I/σ(I) obs: 5.7 / % possible all: 43.6 |
| Reflection | *PLUS Num. measured all: 48145 |
| Reflection shell | *PLUS % possible obs: 43.6 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2BRD Resolution: 2.9→10 Å / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 58.2 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→3.03 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.9 Å / Lowest resolution: 10 Å / σ(F): 0 / % reflection Rfree: 3 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 58.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.9 Å / Rfactor Rfree: 0.297 / % reflection Rfree: 3 % / Rfactor Rwork: 0.214 |
