 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-20027 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Human apoferritin at 2.32 Angstrom | ||||||||||||
 マップデータ マップデータ | Human Apoferritin | ||||||||||||
 試料 試料 |
| ||||||||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | ||||||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 2.32 Å | ||||||||||||
 データ登録者 データ登録者 | Zhang K / Pintilie G / Li S / Chiu W | ||||||||||||
| 資金援助 |  米国, 3件 米国, 3件
| ||||||||||||
 引用 引用 | ジャーナル: Nat Methods / 年: 2020 タイトル: Measurement of atom resolvability in cryo-EM maps with Q-scores. 著者: Grigore Pintilie / Kaiming Zhang / Zhaoming Su / Shanshan Li / Michael F Schmid / Wah Chiu / 要旨: Cryogenic electron microscopy (cryo-EM) maps are now at the point where resolvability of individual atoms can be achieved. However, resolvability is not necessarily uniform throughout the map. We ...Cryogenic electron microscopy (cryo-EM) maps are now at the point where resolvability of individual atoms can be achieved. However, resolvability is not necessarily uniform throughout the map. We introduce a quantitative parameter to characterize the resolvability of individual atoms in cryo-EM maps, the map Q-score. Q-scores can be calculated for atoms in proteins, nucleic acids, water, ligands and other solvent atoms, using models fitted to or derived from cryo-EM maps. Q-scores can also be averaged to represent larger features such as entire residues and nucleotides. Averaged over entire models, Q-scores correlate very well with the estimated resolution of cryo-EM maps for both protein and RNA. Assuming the models they are calculated from are well fitted to the map, Q-scores can be used as a measure of resolvability in cryo-EM maps at various scales, from entire macromolecules down to individual atoms. Q-score analysis of multiple cryo-EM maps of the same proteins derived from different laboratories confirms the reproducibility of structural features from side chains down to water and ion atoms. | ||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_20027.map.gz | 135.5 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-20027-v30.xmlemd-20027.xml | 19.6 KB 19.6 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  emd_20027.png emd_20027.png | 194.4 KB | ||
| その他 | emd_20027_additional_1.map.gzemd_20027_additional_2.map.gzemd_20027_half_map_1.map.gzemd_20027_half_map_2.map.gz | 108.6 MB 837.3 KB 109.6 MB 109.7 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-20027ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20027 http://ftp.pdbj.org/pub/emdb/structures/EMD-20027ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20027 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_20027_validation.pdf.gz | 77.9 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_20027_full_validation.pdf.gz | 77 KB | 表示 | |
| XML形式データ | emd_20027_validation.xml.gz | 493 B | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-20027ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-20027 | HTTPS FTP |
-関連構造データ
| 関連構造データ | C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ |






















-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_20027.map.gz / 形式: CCP4 / 大きさ: 144.7 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Human Apoferritin | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
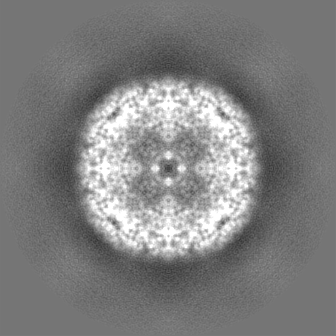
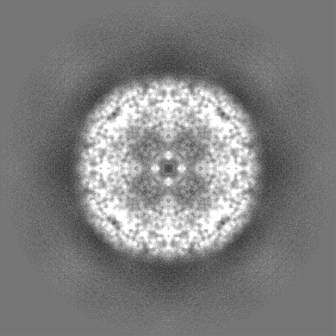
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 0.65 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-添付データ
-追加マップ: unmasked map
| ファイル | emd_20027_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | unmasked map | ||||||||||||
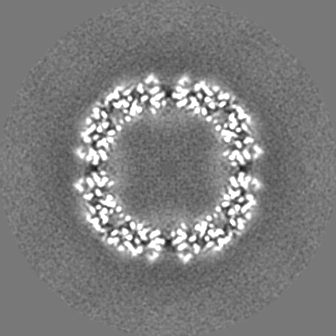
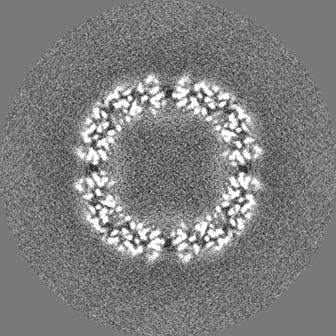
| 投影像・断面図 |
| ||||||||||||
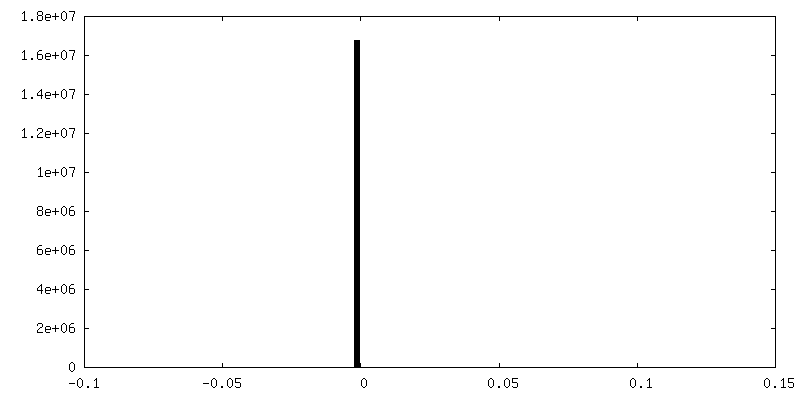
| 密度ヒストグラム |





-追加マップ: one protomer
| ファイル | emd_20027_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | one protomer | ||||||||||||
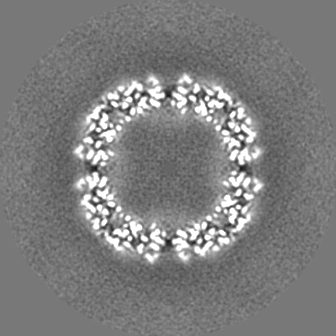
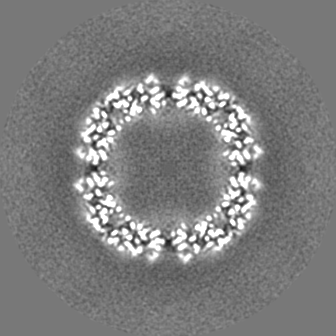
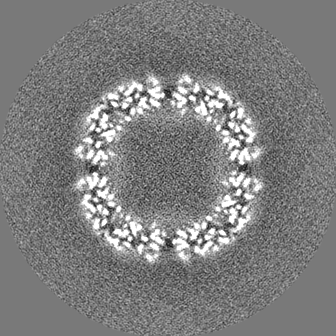
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |







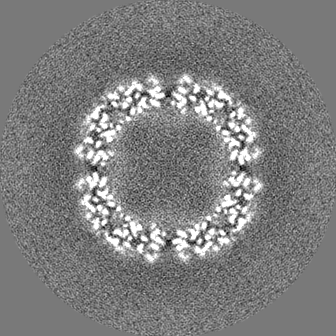
-ハーフマップ: half map 1
| ファイル | emd_20027_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | half map 1 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
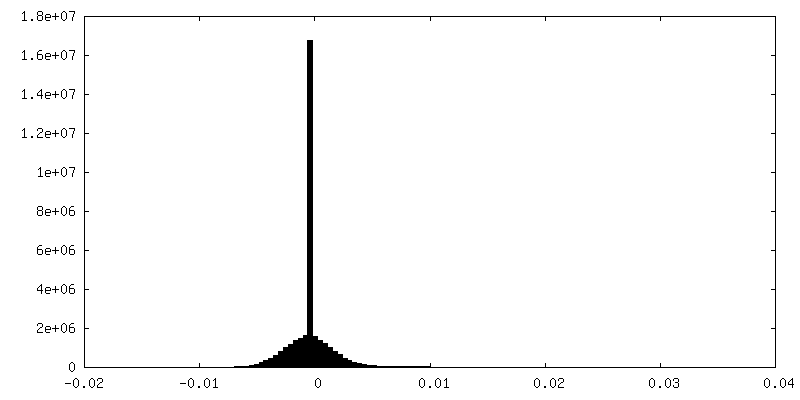
| 密度ヒストグラム |
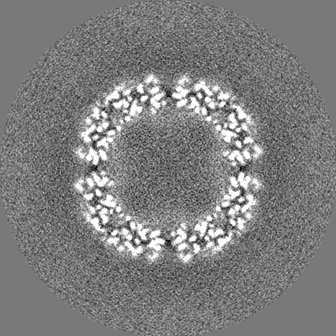
-ハーフマップ: half map 2
| ファイル | emd_20027_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | half map 2 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
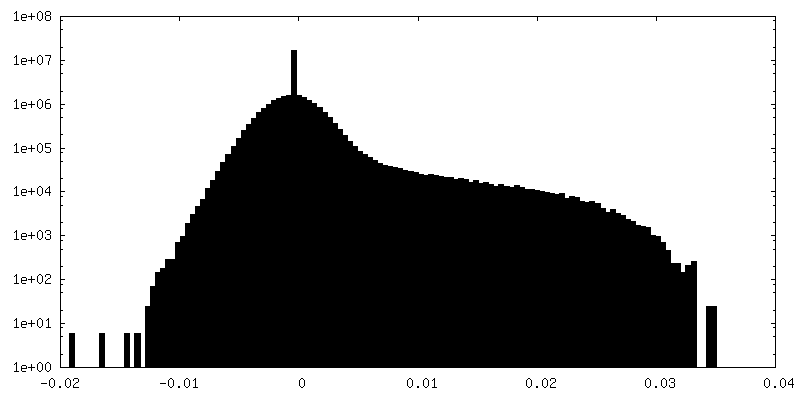
| 密度ヒストグラム |

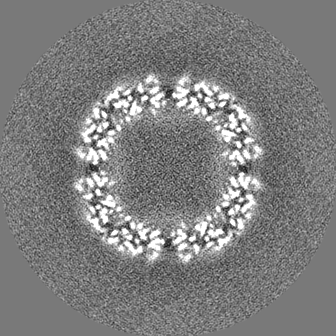
- 試料の構成要素
試料の構成要素
-全体 : Human Apoferritin
| 全体 | 名称: Human Apoferritin |
|---|---|
| 要素 |
|
-超分子 #1: Human Apoferritin
| 超分子 | 名称: Human Apoferritin / タイプ: complex / ID: 1 / 親要素: 0 |
|---|---|
| 由来(天然) | 生物種: Homo sapiens (ヒト) |
| 組換発現 | 生物種:  |
| 分子量 | 実験値: 480 KDa |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 1.5 mg/mL |
|---|---|
| 緩衝液 | pH: 8 |
| グリッド | 詳細: unspecified |
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / チャンバー内温度: 277.15 K / 装置: FEI VITROBOT MARK IV |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: COUNTING / デジタル化 - 画像ごとのフレーム数: 1-30 / 撮影したグリッド数: 1 / 実像数: 1100 / 平均露光時間: 6.0 sec. / 平均電子線量: 7.0 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | C2レンズ絞り径: 70.0 µm / 最大 デフォーカス(補正後): 1.5 µm / 最小 デフォーカス(補正後): 0.4 µm / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.7 mm / 倍率(公称値): 215000 |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
