

SeSAW Tutorial
※旧版ヘルプページはこちら
このチュートリアルではPDBjのサービスの1つとして提供されている SeSAW について解説しています。
用いている手法など基本的なことについては SeSAWに関する文書 をご覧下さい。
[PDB IDによる構造指定]
下図には、SeSAWのトップページでPDB IDを指定する方法を示しています。

結果を電子メールで受け取りたい場合はアドレスを入力して下さい。 結果をウェブブラウザで得ることもできます。
注意点は以下の通りです:
- 鎖ID(chain ID)は必ず入力して下さい。鎖IDが分からない場合はあらかじめ確認して下さい。
- 有効なPDBエントリーだけが処理されます。指定しようとしているPDB IDが確かに存在するかあらかじめ確認しておくのがいいでしょう。
- 条件内容によっては結果が返るまでに1時間かかることもありますが、結果が出るまで我慢して待って下さい。
- 結果が返らなかった場合、または結果が変に思えた場合は、sesaw@protein.osaka-u.ac.jp までメールで知らせて下さい。
[ファイル送信による構造指定]
実験的に決定された構造の場合
下図には、SeSAWのトップページでPDBフォーマットのファイルを指定する方法を示しています。
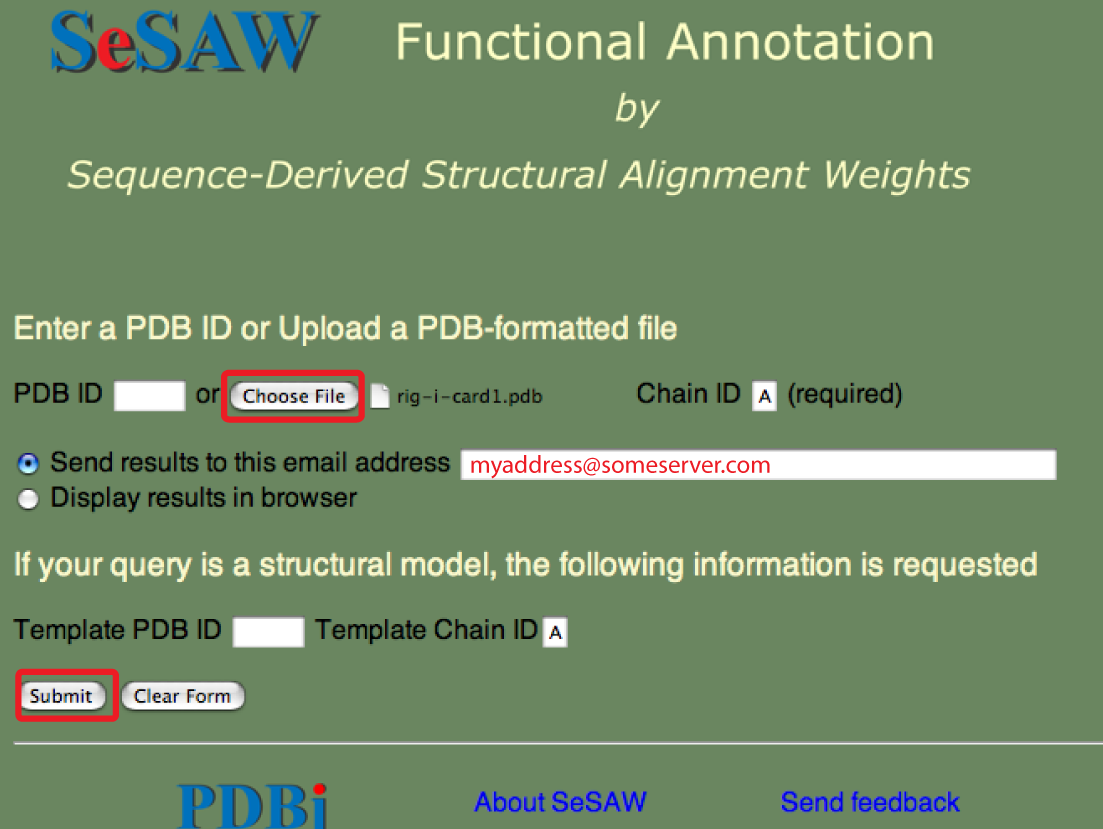
先ほどと同様、 結果を電子メールで受け取りたい場合はアドレスを入力して下さい。 結果をウェブブラウザで得ることもできます。
なお、ファイルで条件指定を行う場合は、先ほどの場合に比べより時間がかかる傾向があることに注意して下さい。 なぜなら、PDB IDで指定する場合は、あらかじめ処理を行っており、本処理時の作業内容を減らしているからです。
構造モデルの場合
構造モデル情報を用いる場合も手順は同じですが、 鋳型となるPDB IDを指定しておくことをお勧めします。 なぜなら、鋳型分子と構造的に似ているものは素早く探し出すことができるからです。 これによって処理に要する時間を大幅に短縮することができます。
[実験的に決定された構造の例]
2czl
2czl はタンパク3000プロジェクトの中で解明された構造ゲノミクス(SG)対象の1つです。 2czlを使ったSeSAWクエリの実行方法は以下の通りです。
1. PDB ID(2czl)、鎖ID(A)、電子メールアドレスを上記のように入力します。
2. しばらく待つと次のようなメールが送られてきます。
From: sesaw@protein.osaka-u.ac.jp Subject: SeSAW Query 2czlA Date: October 7, 2008 5:48:15 PM JST To: xxxx Thank you for using the SeSAW functional annotation server. The results of your query will be stored for 2 weeks at the following URL: http://pdbjc01.protein.osaka-u.ac.jp:8888/tmp/SS17645/2czlA_top.html We hope these results will be useful for your research. Best wishes, -SeSAW
もし1、2時間経ってもメールが来なかった時は、何か間違った指定があったのでしょう。 その時は我々にメールして下さい。メールアドレスは sesaw@protein.osaka-u.ac.jp です。
3. メールに記載されたアドレスにアクセスすると下図のようなページが表示されます。

2czlAの2つのドメインだけでなく、鎖全体の結果もあることに注目して下さい。 SeSAW は可能な限り指定されたクエリをドメインに分解して処理します。 なぜならこれはスコアに大きく影響するからです。
4. 鎖全体へのリンクをクリックして下さい。以下に示すような結果一覧が表示されます。
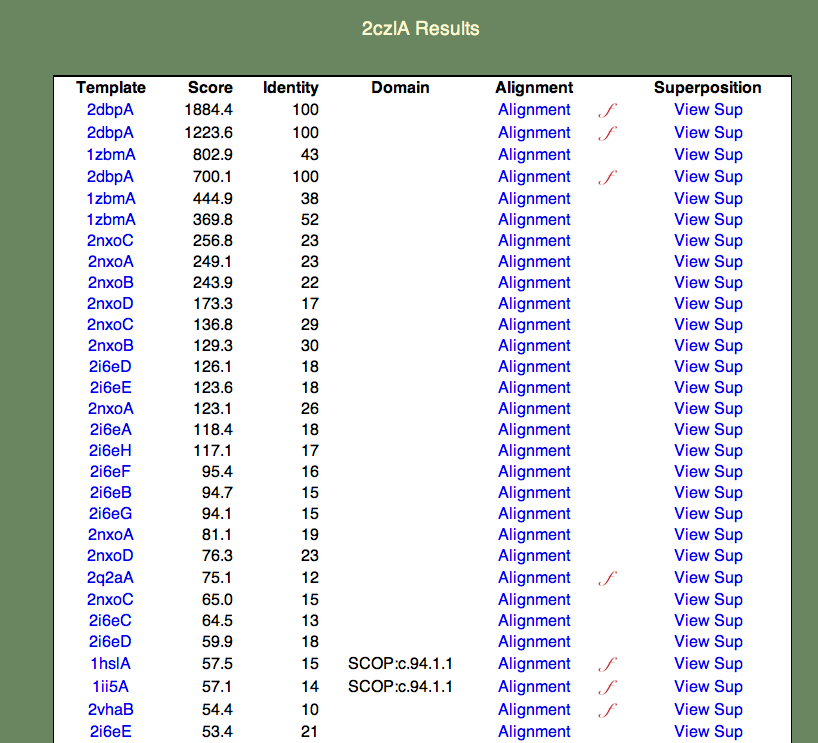
表の形式は以下の通りです。
- 「Templete」列には 2czl と似ているPDBエントリーの PDBj Mine (PDBエントリー検索結果)へのリンクが、IDの小さい順(降順)で表示されます。 なお、1つのエントリーが複数現れることがあります。これは鋳型をドメインに分解したものも処理していることによります。表記を簡潔にするため、ドメイン番号は結果ページに表示していません。
- 「Score」列には SeSAW スコアが表示されます。 スコアと特定の予測の信頼性との関係に関する議論が こちら に掲載されています。
- 「Identity」列には、対応する残基の組み合わせがどれだけ一致しているかを示しています(ギャップは含まれません)
- 「Domain」列には、 SCOP や CATH のIDが表示されます(あった場合)。
- 「Alignment」列には、説明付加されたJalview配列へのリンクが表示されます。 右に赤色の筆記体fが表示されていた場合、配列に関する機能説明があることを示します。
- 「Superposition」列には、 jV 分子構造閲覧ソフトによる ASH 構造重ね合わせ画像ページへのリンクが表示されます。
5. 次に 2czl の機能に対する一般的情報を取得します。 Template列を上から見ていくと、上25件程度の鋳型は全てSG対象であることが分かるでしょう。 つまり、それらには詳細な機能説明がなく、ほとんどの場合は1次文献参照がないということです。 ところが、更に下に見ていくと、文献参照がある最も上にある鋳型は全てアミノ酸結合蛋白質であることが分かるでしょう。
6. 次に特定のクエリー鋳型配列を調べてみましょう。 鋳型 1ii5A を含む行を探して下さい。 alignment リンクをクリックすると、下図のようなページが表示されるでしょう。
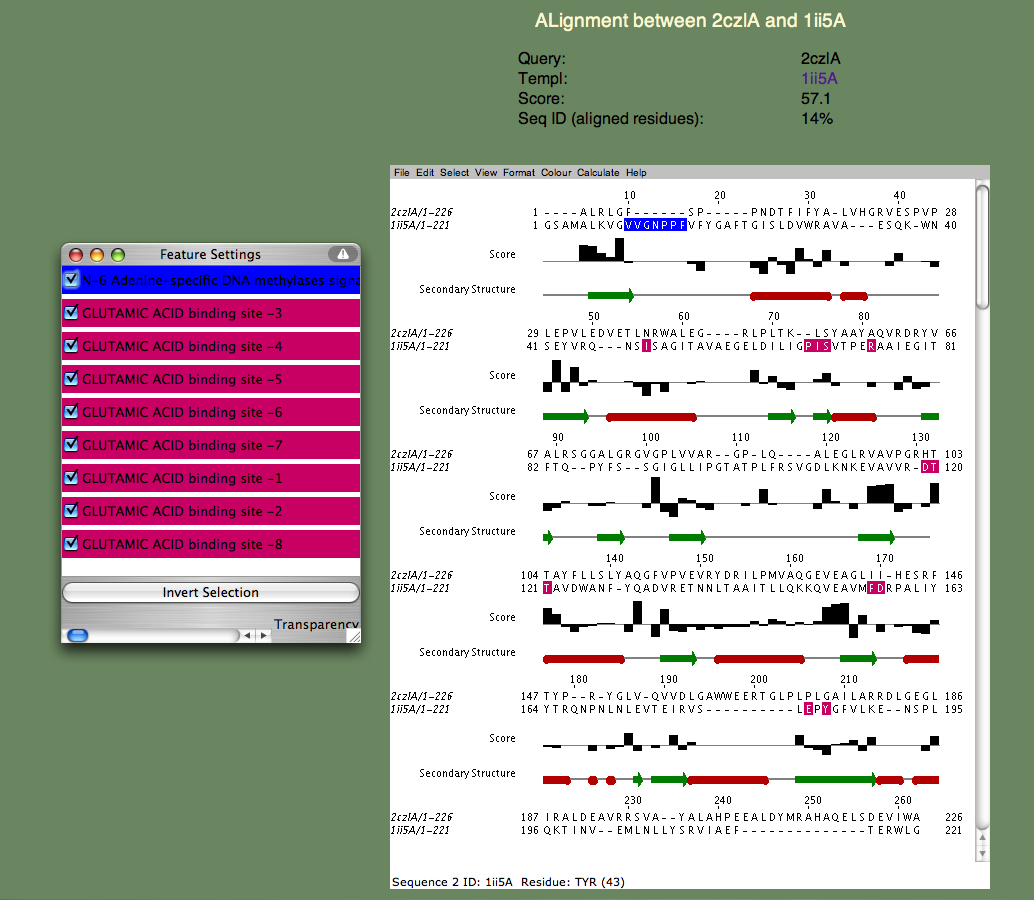
もしこのようなページが表示されず、代わりに X が表示された時は、ブラウザ上でJalviewアップレットの動作に何らかの問題が発生しています。 その時は Jalview のホームページ を確認すると何か役立つ情報が得られるでしょう。 特に「News Mailing List」を確認して下さい。 Jalview の関係者はよく助けてくれて、素早く質問に答えてくれます。 配列ウインドウには指定条件(クエリ)に対するより詳細な説明が表示されます。 ポップアップする「Features」ウインドウには機能説明がある連続配列モチーフの一覧が表示されます。一番最初には「N-6 Adenine-specific DNA methylases signature」(N-6アデニン特異的DNAメチル化酵素署名)が青色で表示されます。 鋳型中にあるこの配列断片は配列比較におけるギャップに相当します。 言い換えると、2czlAはquestionの中に特徴を持っていません。
次に、残りの特徴を見てみましょう。 全て 'GLUTAMIC ACID binding site'(グルタミン酸結合部位)に関するもので、赤紫色で表示されています。 これらの残基は配列全体に広がり、そのほとんど(プロリンを除く)はギャップ部分には並んでいません。 残基の1文字記号の下には棒グラフが表示されます。 これは残基ごとのSeSAWスコアを示していて、これら残基のうちの3つ(S57、T106、T107)は好ましいが、他よりはマイナスです。 このことは結合部位の位置が恐らく2czlの異なる機能部位に関係しているであることを示しています。 またおもしろいことに、1ii5Aよりも高いスコアを示し、より塩基性のアミノ酸(アルギニン)に結合する2q2aAを鋳型として選択すると、2つのスレオニン(T106、T107)は保存されているが、セリン(S57)と残りの残基は保存されていないことが分かります。 これらの結果から考えると、結合部位らしい場所は、正確なリガンドでなくても予測することができます。
実際、2czlの構造は酒石酸(tartaric acid)と一緒に結晶化されたものです。 そのため、酒石酸の場所を見ると、1ii5A と 2q2aA アミノ酸の場所と合致していることが分かります。 そのことを見るには、2dbpAテンプレート(一覧の最初)をクリックして下さい。 そうすると、リガンドが結合する可能性のある場所についてより確信を持ち、結合しうるリガンドに酒石酸が含まれていることが分かります。 しかし、ここにある情報だけでは 2czlA の機能が何であるのかについては分かりません。
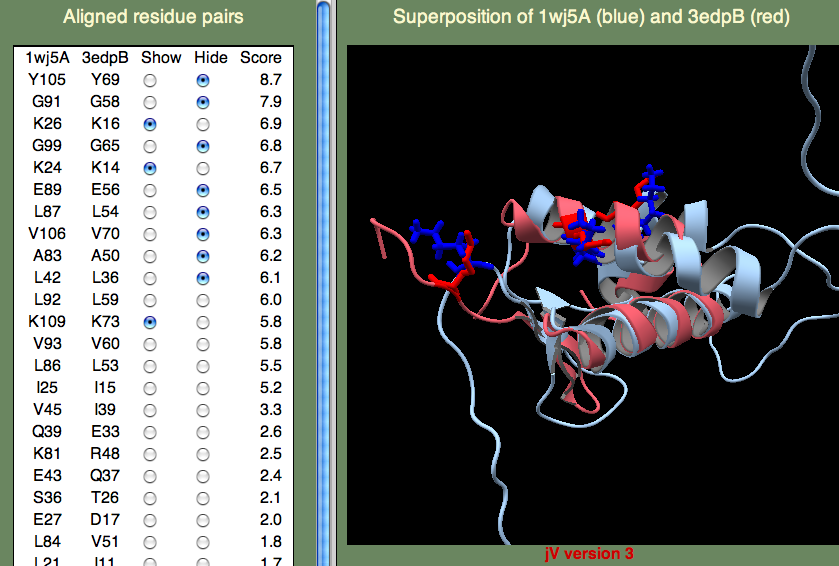
7. では次に構造重ね合わせの特徴について見てみましょう。 1ii5 の行にある 'View Sup' リンクをクリックすると、中にjVウインドウがある新しいページが表示されます。 最初にこのリンクをクリックした時は、下記のようなウインドウが表示され、電子証明書を承認するか確認を求められます。
「Trust」(信頼)ボタンをクリックして承認すれば、下図のような構造重ね合わせの結果が表示されます。
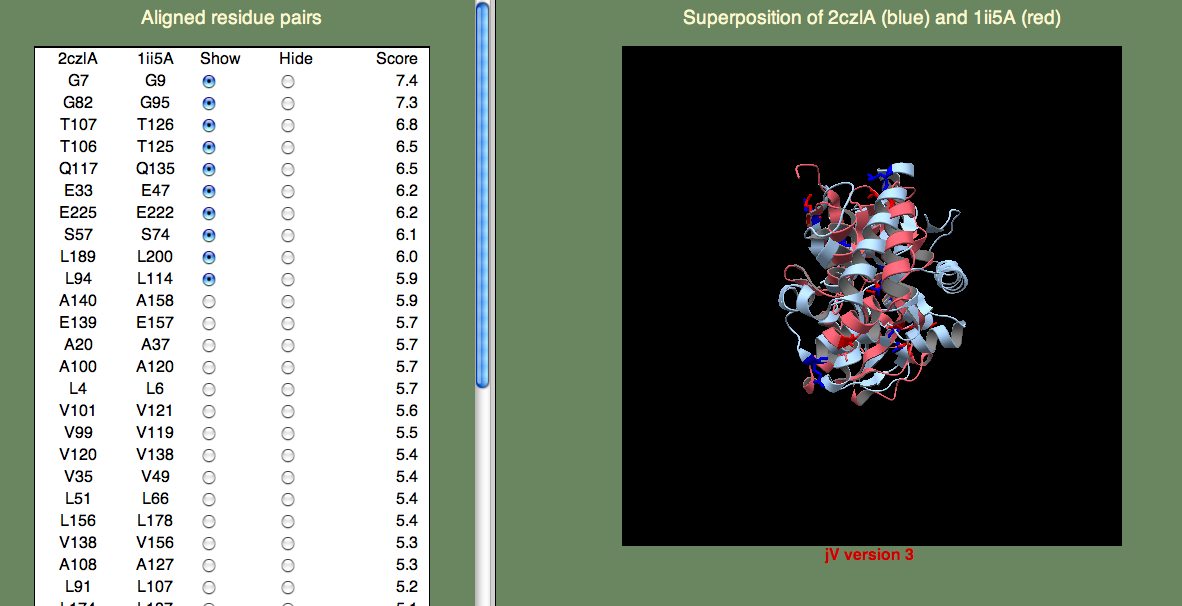
左側のパネルの Show/Hide ラジオボタンを使って、対となる残基(残基ペア)を表示したり非表示にしたりすることができます。 これらの対はSeSAWスコアの大きいものから順に表示されています。初期状態では、上位10組の残基が選択されています。
保存されたスレオニン(T106、T107)とセリン(S57)を見ると、これらの残基が確かにクラスターを形成していることが分かります。 他にどの高いSeSAWスコアの残基ペアが、このクラスターに属しているでしょうか? その数個の残基ペアを見つけるにはかなりの回数クリックしないといけないかもしれませんが、 A108 もこのクラスターの一部分です。 では、G82 についてはどうでしょうか? この残基はかなり遠くに離れていますが、ここで説明するように結合部位を開いてリガンドがアクセスできるようにするのに重要であることが分かっています。 そして、幸いなことにこの残基ペアが高いスコアで一覧に表示されていることでそのことが分かるのです。
他の残基ペアを選んで他のクラスターでも試してみることができます。 例えば、L4、L94、V99、A100、V101、Q117、V120、E139 で構成されるクラスターは1つのドメインの表面に位置しています。 この部分は何か意味を持っていると考えたくなりますが、 個々のドメイン(この場合 2czlA_Dom-2)を調べることでより詳しく見ることができます。
1wjk
では、スコアが上位に位置する配列の分析を直接進めることにします。 結果の表を以下に示します。
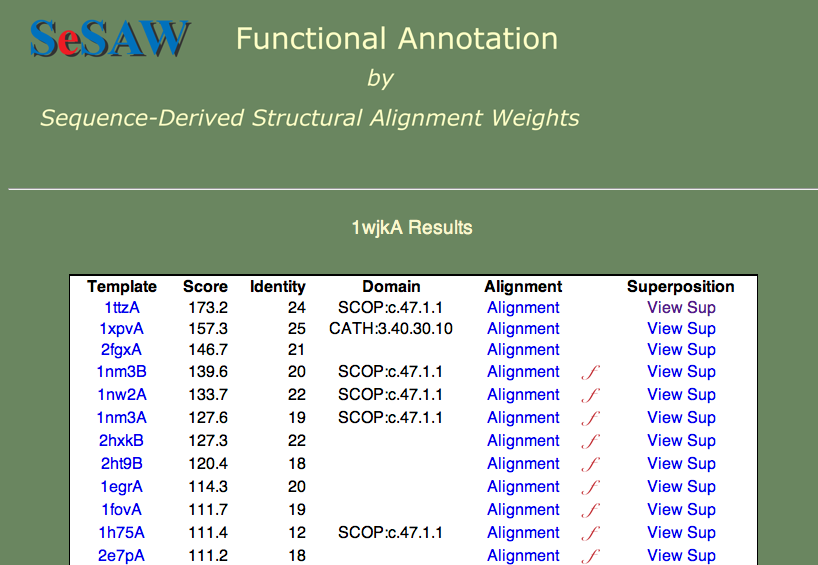
答えが欲しい質問は次の2つです。
1)1wjk の機能は何なのか?
2)どの残基が機能に重要そうであるのか?
最初の質問に答えるために、スコアが上位の鋳型はどんな機能を持っているのか見てみましょう。
機能が分かっていないもの(1ttza、1xpvA、2fgxA)からグルタレドキシン(glutaredoxin、1nm3B)やチオレドキシン(thioredoxin、1nw2A)まで様々なものが見つかります。
これらの蛋白質は全て同じスーパーファミリーの一部なので、ここで知りたい蛋白質も同じグループに属しているだろうということが言えます。
次に、2つ目の質問に答えるために、チオレドキシンとグルタレドキシンが何をしているのか見てみましょう。 どちらも酸化還元反応(oxidation/reduction (redox) reaction)を通して信号伝達に関わっている蛋白質で、 この反応は、酸化する(対を解消する)ことも還元する(ジスルフィド結合を作り対になる)こともできるシステインの対によって促進されます。 この2つの蛋白質はどの分子に作用するかという点で見れば違うものです。 チオレドキシンはチオレドキシン還元酵素(thioredoxin reductase)によって還元される大きい方の蛋白質です。 グルタレドキシンはグルタチオン(glutathione)によって還元される小さい方の蛋白質です。 だから、還元中心を特定するにはシステインの対を見つければよいということになります。 そこで、蛋白質間相互作用部位(グルタレドキシン)や小分子結合部位(チオレドキシン)と考えられる場所を探します。 システインは簡単に見つけられるでしょう。なぜなら、それは最も高いスコアを持っているからです。 次に、他に良いスコアの残基がどれなのかを見てみましょう。 一番良いスコアの鋳型 1ttzA(注釈なし)を見ると、スコアの良い多くの疎水性残基があるのが分かるでしょう(下図参照)。
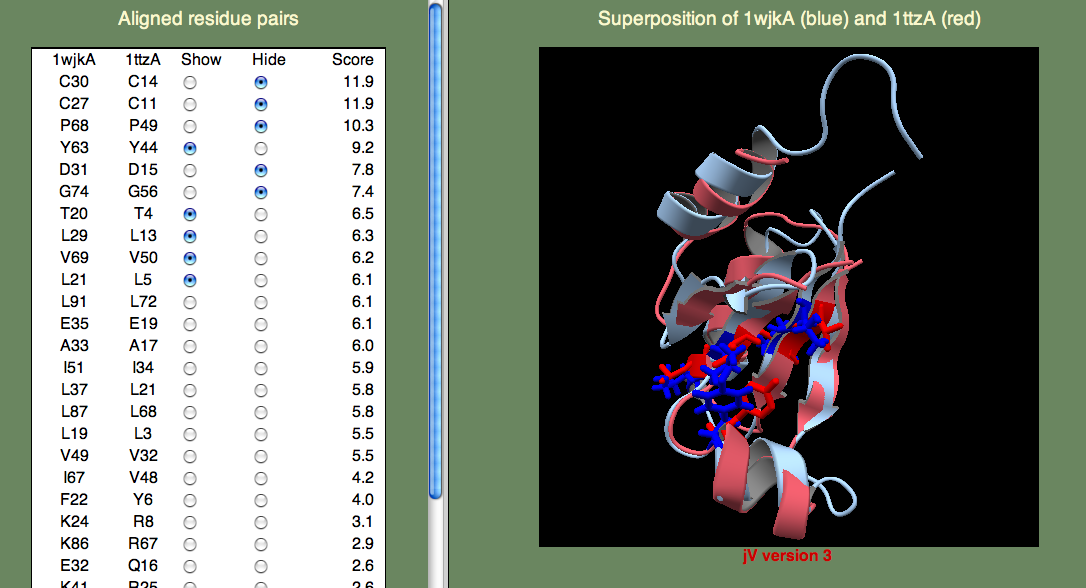
これらの残基は折りたたみを安定化させる疎水性中心の一部にすぎないのかもしれません。 あるいは、蛋白質間結合部位の一部なのかもしれません。 このデータだけでは確かなことは言えませんが、 これら蛋白質を代表するスーパーファミリーは構造ゲノミクスの取り組みによって急速に増えていて、 近い未来にこれらの機能についてより明らかになって行くことでしょう。
1wj5
ここで再び、蛋白質の一般的な機能を知り、重要である可能性のある残基を特定してみることにします。 スコアが上位のものが以下のように表示されます。
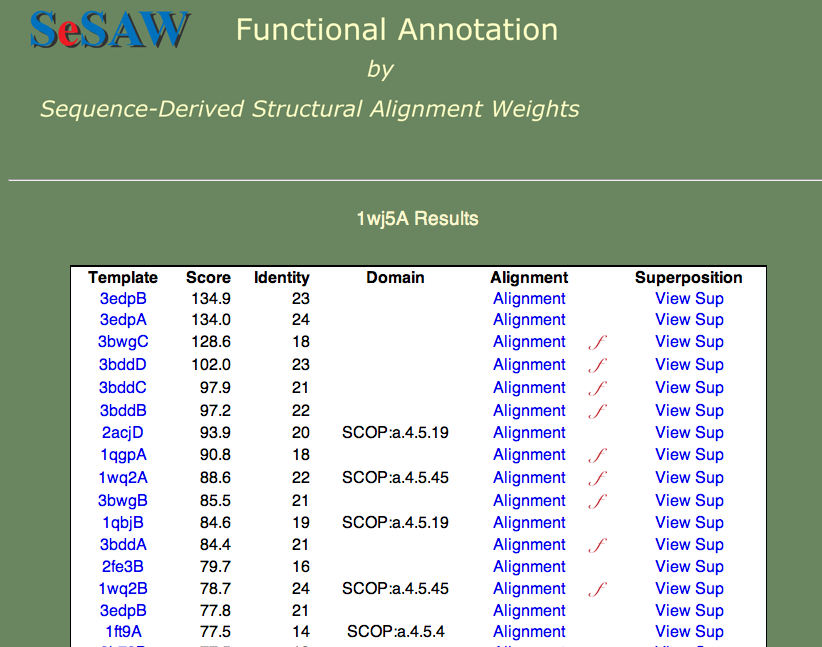
基本的な機能を推測できますか? この例も難しい場合の1つです。 なぜなら、答えはまだ分かっていないからです。 しかし、各鋳型を見るとヌクレオチド結合が共通のテーマであることが確認できます。 更に、最初の注釈のある鋳型(3bwg)への構造比較をみて、 1wj5の機能部位は鋳型の機能部位と一致するかどうかを確認することができます(下図参照)。
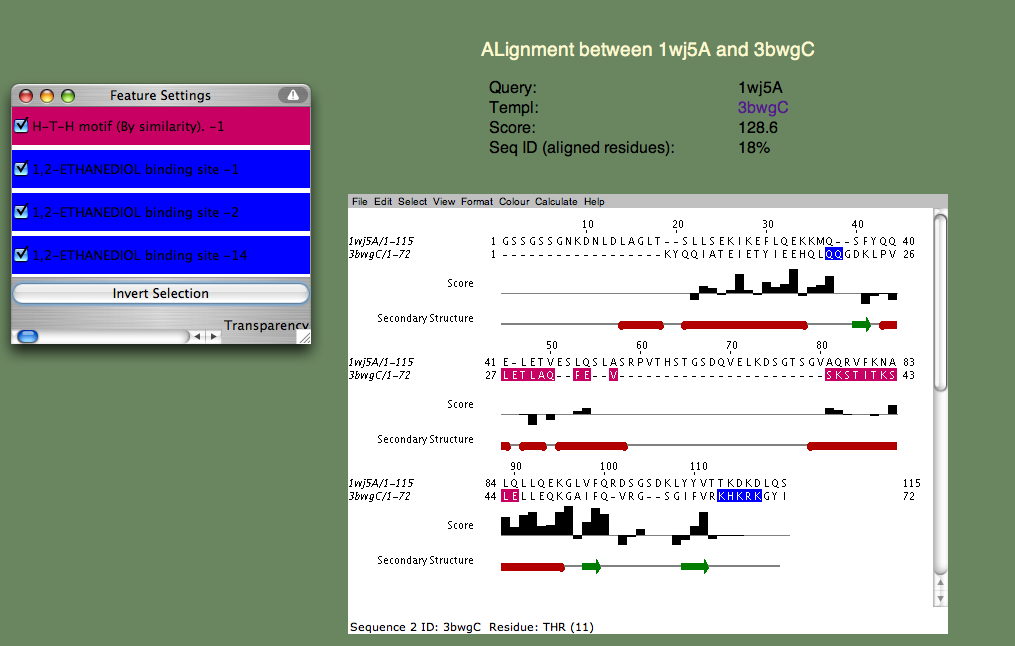
H-T-H は「へリックス-ターン-へリックス モチーフ」(Helix-Turn-Helix motif)を示していて、名前が示すように、これは配列モチーフというよりは構造モチーフです。 更に、配列比較した領域(ピンク)はあまり多くありません。 2つ目のモチーフ(1,2-エタンジオール結合部位)は反対側にあります。 これは構造的にあまり重なりませんが、類似した配列が多く見られます(Q35とC末端のリジン残基)。 これらの残基は、重ならない配列「SRPVTHSTGSDQVELKDSGTSGV」と並んでいて、1wj5のdisorder(構造不確定)部分を補完している。 disorder領域は転写制御によく関わっていることが知られていて、 この場合もこれに当てはまると考えたくなります。 恐らく1wj5についての最善の答えは、ヌクレオチド相互作用に関係していそうである、 というもので、この答えは最もスコアが高く、注釈がなされていない鋳型3edpBを調べた結果に由来します。 構造重ね合わせによって3つの塩基性残基(K26、K24、K109)のスコアが良く、ある程度ひとかたまりになっています。 これはまだかなり不確定な話ですが、恐らくヌクレオチド結合部位を最も良く推測しているでしょう。
[構造モデルの例]
RIG-I のヘリカーゼドメイン
1. まず、モデルを作らなければなりません。 ここに示すものはそれ自体が芸術です。したがって、ここでは深く取り上げることはしません。 SWISSモデルなどモデル構築に関する素晴らしい情報源があります。 この例では、HHpredサーバを使ってレチノイン酸(retinoic acid)誘導性遺伝子I(RIG-I)にあるヘリカーゼドメイン(helicase domain)のモデルを作りました。 まずUniProtで「mouse」と「rig-i」のキーワードで検索してマウスのRIG-I遺伝子を見つけ、登録IDをクリックし、要約ページの右上にあるFASTAを選択して配列全体をダウンロードしました。 FASTA配列とは下記ようなものです。
>sp|Q6Q899|DDX58_MOUSE Probable ATP-dependent RNA helicase DDX58 OS=Mus musculus GN=Ddx58 PE=2 SV=1 MTAAQRQNLQAFRDYIKKILDPTYILSYMSSWLEDEEVQYIQAEKNNKGPMEAASLFLQY LLKLQSEGWFQAFLDALYHAGYCGLCEAIESWDFQKIEKLEEHRLLLRRLEPEFKATVDP NDILSELSECLINQECEEIRQIRDTKGRMAGAEKMAECLIRSDKENWPKVLQLALEKDNS KFSELWIVDKGFKRAESKADEDDGAEASSIQIFIQEEPECQNLSQNPGPPSEASSNNLHS PLKPRNYQLELALPAKKGKNTIICAPTGCGKTFVSLLICEHRLKKFPCGQKGKVVFFANQ IPVYEQQATVFSRYFERLGYNIASISGATSDSVSVQHIIEDNDIIILTPQILVNNLNNGA IPSLSVFTLMIFDECHNTSKNHPYNQIMFRYLDHKLGESRDPLPQVVGLTASVGVGDAKT AEEAMQHICKLCAALDASVIATVRDNVAELEQVVYKPQKISRKVASRTSNTFKCIISQLM KETEKLAKDVSEELGKLFQIQNREFGTQKYEQWIVGVHKACSVFQMADKEEESRVCKALF LYTSHLRKYNDALIISEDAQMTDALNYLKAFFHDVREAAFDETERELTRRFEEKLEELEK VSRGPSNENPKLRDLYLVLQEEYHLKPETKTILFVKTRALVDALKKWIEENPALSFLKPG ILTGRGRTNRATGMTLPAQKCVLEAFRASGDNNILIATSVADEGIDIAECNLVILYEYVG NVIKMIQTRGRGRARDSKCFLLTSSADVIEKEKANMIKEKIMNESILRLQTWDEMKFGKT VHRIQVNEKLLRDSQHKPQPVPDKENKKLLCGKCKNFACYTADIRVVETSHYTVLGDAFK ERFVCKPHPKPKIYDNFEKKAKIFCAKQNCSHDWGIFVRYKTFEIPVIKIESFVVEDIVS GVQNRHSKWKDFHFERIQFDPAEMSV
そして、この配列を初期設定のままHHpredに送信すると、次のような結果ページが表示されるでしょう。
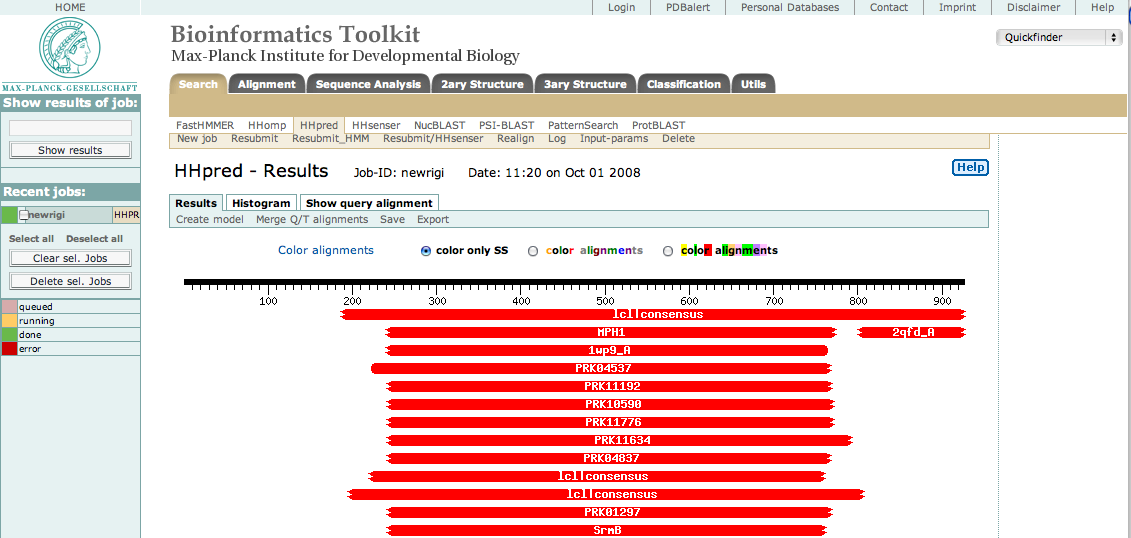
図に示したように、250から775辺りの中央領域にたくさんヒットします。 これはヘリカーゼドメインです。 送信した配列の中でこれらの残基だけを選択して再度送信し、 最もよくヒットしたものを選択します。 すると次のような断片で構成される3つのサブドメインを特定することができました。
>ATPase-like 1 ECQNLSQNPGPPSEASSNNLHSPLKPRNYQLELALPAKKGKNTIICAPTGCGKTFVSLLICEHRLKKFPCGQKGKVVFFANQIPVYEQQAT VFSRYFERLGYNIASISGATSDSVSVQHIIEDNDIIILTPQILVNNLNNGAIPSLSVFTLMIFDECHNTSKNHPYNQIMFRYLDHKLGESRDPL PQVVGLTAS >Helical VGVGDAKTAEEAMQHICKLCAALDASVIATVRDNVAELEQVVYKPQKISRKVASRTSNTFKCIISQLMKETEKLAKDVSEELGKLFQIQN REFGTQKYEQWIVGVHKACSVFQMADKEEESRVCKALFLYTSHLRKYNDALIISEDAQMTDALNYLKAFFHDVREAAFDETERELTRRF EEKLEELEKVS > ATPase-like 2 RGPSNENPKLRDLYLVLQEEYHLKPETKTILFVKTRALVDALKKWIEENPALSFLKPGILTGRGRTNRATGMTLPAQKCVLEAFRASGDNNI LIATSVADEGIDIAECNLVILYEYVGNVIKMIQTRGRGRARDSKCFLL
次の指示は2008年9月に得られたHHpredサーバの結果に基づいています。この結果は変わることがあります。
次にページの一番上にある「Create model」ボタンを選択し、鋳型としてPDBエントリー2db3を選択して、2つ目の「ATPアーゼ様ドメイン」(ATPase-like domain)のモデルを作ります。 この鋳型は初期状態で選択されている鋳型ではないことに注意して下さい。 これを選択したのは1wp9(19%)よりも配列類似度が高かった(25%)からです。 実際この指示を行うにはMODELLERライセンスキーを取得する必要がありますが、 その結果作られるモデルを こちら から取得できます。
ここで一つ厄介なことがあることに注意して下さい。
MODELLER は初期状態ではPDBファイルに鎖ID(chain ID)を付加してくれません。 一方、SeSAWでは鎖IDが必要です。 この問題を回避するために、PDBフォーマットファイルに鎖IDを付加するウェブベースのユーティリティを用意しました。ユーティリティには こちら からアクセスできます。 このユーティリティを使ってPDBフォーマットファイルに鎖IDを付加して下さい。
2. 前述のモデル構造をSeSAWに送信します。 鋳型となる構造のPDB IDを入力するのを忘れないで下さい(この例の場合 2db3)。 ここでは送信過程が成功したものとして結果の分析を省略します。
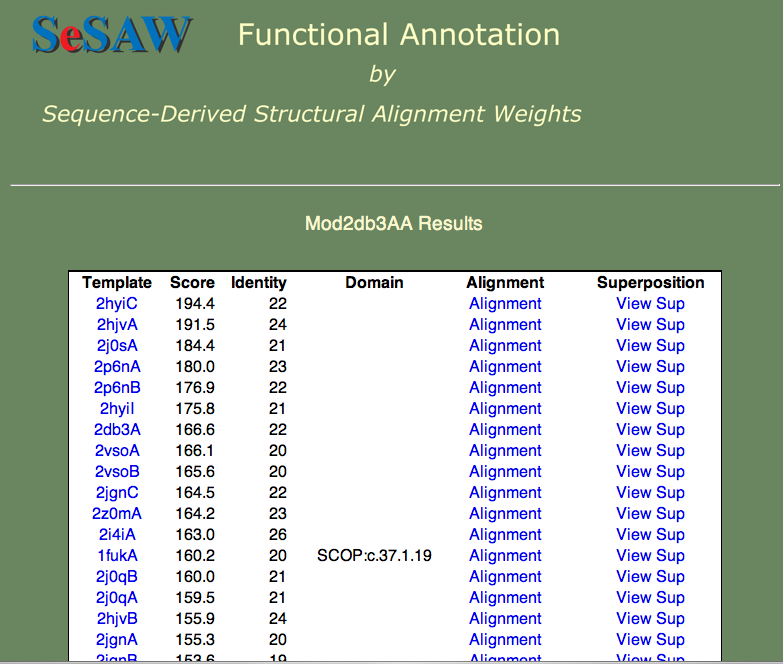
3. 2db3 の結果表は以下のようになります。
上位にヒットしているのは全てヘリカーゼであることから、指定したモデルもそうであるとある程度確信することができます。
ヘリカーゼはほどけた2本鎖ヌクレオチド(DNAまたはRNA)を安定化させます。 ヌクレオチドは負に帯電しているという事実から、ヌクレオチド結合部位には正に帯電したアミノ酸(アルギニン(R,arginine)、リジン(K,lysine)、ヒスチジン(H,histidine))があるものと期待できます。 いくつかのスコアが高い比較の中の高いスコアの残基を見て、結合部位の候補となる残基を見つけることができますか? 下には「エキソンジャンクション複合体(exon junction complex)とそれに捕捉された DEADボックスヘリカーゼ(DEAD-box helicase)」(PDB ID 2hyi)の比較結果を示しています。

モデルの最後20残基を見ると、4つのアルギニン残基と1個のリジン残基があるのが分かるでしょう。 このうち R 116は多くの配列中に見られます。 R 116の近くにある他の2残基はどうでしょうか? ヒント:「近い」というのは必ずしも残基番号が近いことを意味している訳ではありません。 「近い」残基を見つけるには、構造重ね合わせを見ればいいのです。
注目している特定の領域において配列と二次構造に少なくとも1つのギャップがはさまっていることは必ずしも普通のことではない、ということにも注意してして下さい。 (例えば、ループとへリックスの区別は明確ではありません) 言い換えると、この注目している領域はモデルにとってはそれほど重要ではないということです。 だから、データの機能的解釈には注意しなければいけません。 なぜなのかその理由を思いつきますか? 結合部位は機能を果たすために柔軟でなければならないことがよくあります。 例えば、蛋白質が結合状態と非結合状態で異なる立体構造を取るというのはよくあることです。 ここにある鋳型は異なる立体構造の集まりを代表したものです。 ここで指示した条件(クエリ)と違ったヌクレオチドとも結合します。 この点で、C末端領域は恐らくRNA結合に関わっているということが言えますが、 RNA認識においてこの領域が果たす正確な役割を突き止めるには、更なる実験的証拠(例えば、変異体分析と直接の構造的証拠)が必要です。
Toll様受容体9 の TIR ドメイン
ヒトのトル様受容体9(human Toll-like receptor 9)から得られたトル/インターロイキン-1受容体(Toll/interleukin-1 receptor、TLR-9 のTIRドメイン)配列を以下に示します。
DAFVVFDKTQSAVADWVYNELRGQLEECRGRWALRLCLEERDWLPGKTLFENLWASVYGSRKTLFVLAHTDRVSGLLRASFLLAQQRLLE DRKDVVVLVILSPDGRRSRYVRLRQRLCRQSVLLWPHQPSGQRSFWAQLGMALTRDNHHFYNRNFCQGPTAE
TLRは自然免疫反応(innate immune response)に関わっていて、 膜貫通らせんが続きC末端に信号伝達ドメインがある受容体ドメインを持っています。 信号伝達ドメインは細胞内にある他の信号伝達蛋白質に結合して、感染が見込まれる際に細胞へ警告しています。 ここではTLR-9のTIRドメインにおけるどの部分が、そのような蛋白質間相互作用に関係しているのかを明らかにすることにします。
ヒトのTLR-1(PDB:1fyv)から配列類似度27%のモデルを作り、前述と同様の既定HHpred手順を行いました。 SeSAWの構造比較において最も高いスコアの残基ペアには、TIRドメインに特徴的なたくさんの保存された疎水性残基(W16、W125、Y110、F135、W136)やプロリン(P45、P103、P126)が含まれています。
<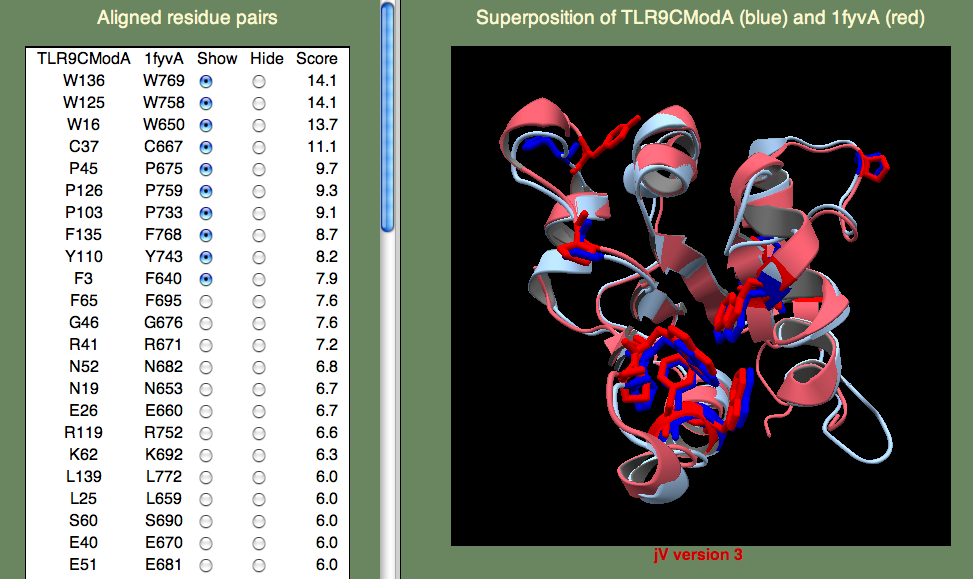
もし PDBエントリー 1fyv の一次参照文献 を見る時間を取れば、 TLR-9とTLR-1、両者から得られたTIRドメインは、受容体信号伝達に関わっていると考えられている「cBBループ」を含む保存パッチにある5つの残基が共通していることが分かるでしょう。 これらの残基(E40、R41、P45、G46、K47)は高いスコアを持っています。 TLR-9とTLR-1のTIR結合部位は同じであると期待してはいないことに注意して下さい。 両者の違いは異なる自然免疫反応受容体によって開始される異なる信号伝達経路に関わっていることと矛盾しないものです。 TLR-9はメチル化されていないCpG DNAを検知し、TLR-1は細菌に由来するリポ蛋白質を検知します。
ブラストシスチスの代替酸化酵素
代替酸化酵素(alternative oxidase,AOX)はミトコンドリアで見られる鉄結合蛋白質です。 ブラストシスチス(Blastocystis)は、酸素を要求しない(嫌気性の)変わったミトコンドリア様の細胞小器官を持つ腸内寄生虫です。 ただこの小器官の機能はまだよく分かっていません。
MFPILSRVFFKREAVVFRGFSVSSYEQFIDKECISKALNKKPNEHYHIFSTRYHSSNREYLTILESCWGEQPKRHPKGVSDRVASGIVNALFKIGN AYFRENYILRAVFLESVASIPGLVCSNLHHLRCLRRLQPDSWIKPLVDEAENERMHLLAVRTYTKLTAVQKLFIRITQFSFVTLFSFLFVFAPRTS HRLVGFLEEHAVDSYTEMIRRIDSNTLENRPATQITKDYWGLPEDATLRDALLVIRADEADHRLVNHSLGDAYDKKTPVSVKKWYAGCAFP VNLHEPFGPYMDFSKYGATKA
知られている配列の類似度は非常に低く、AOXは巧妙な場合です。 ここでいくつかのモデルを試し、既知の機能的に重要な残基との類似比較によって最もよいものを選び出すことにします。 最終的なモデルには鉄結合モチーフが2つ含まれます。 モチーフはE...ExxHというもので、これはグルタミン酸が1つあって、次にさまざまな長さの残基、そして別のグルタミン酸、2つの残基、そしてヒスチジンが来ることを意味します。 これはかなり無原則なようですが、これらの残基は鉄に結合することを忘れないで下さい。 すると、これに近いものが見つかること期待することになります。 また、このモチーフは2回あることも忘れないで下さい。 すると、このパターンに似ていてしかも互いにも似ているそんなものを2つ探すことになります。 時間を節約するために、1つのモデルを こちら 提供します。
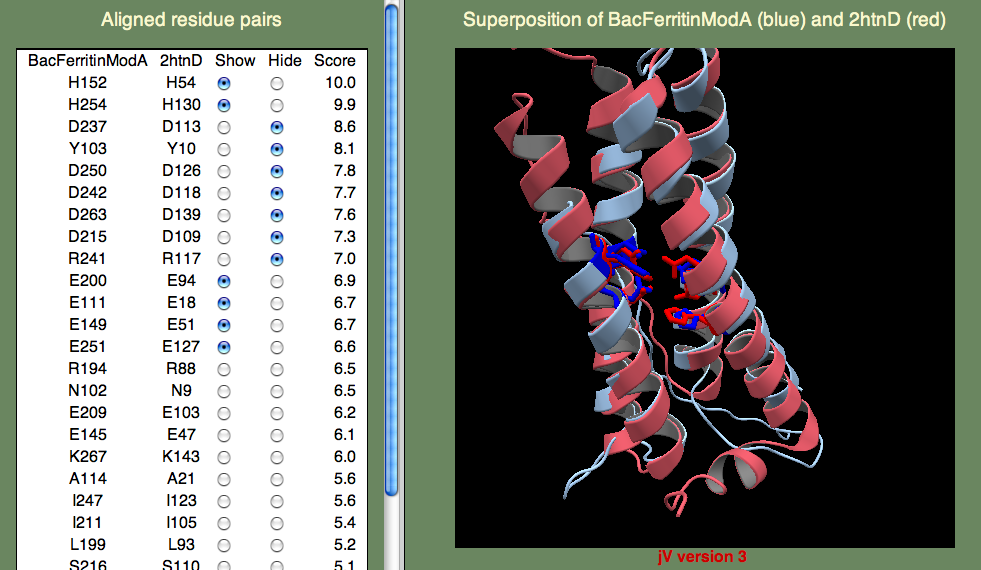
下に示したのはPDBエントリー2htnを鋳型として比較した結果です。 2htnは別の蛋白質(バクテリオロドプシン)であり、AOXと2htnは構造的に一致することは期待していないことを忘れないで下さい。 特にAOXには、バクテリオロドプシンには存在しない膜結合らせんがある点が違っています。 そのらせんがどこなのか推測できますか? ヒントを出しましょう。らせんは膜貫通らせんではなく、膜表面と並行に入って出ているらせんで、膜は通り抜けていません。 この領域を見つけるには、下に示した指定条件の二次構造予測結果を調べなければならないでしょう。




