 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
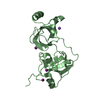
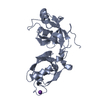
Yorodumi- PDB-3eo6: Crystal structure of protein of unknown function (DUF1255) (AFE_2... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3eo6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of protein of unknown function (DUF1255) (AFE_2634) from ACIDITHIOBACILLUS FERROOXIDANS NCIB8455 at 0.97 A resolution | ||||||
 Components Components | protein of unknown function (DUF1255) | ||||||
 Keywords Keywords |  structural genomics / unknown function / AFE_2634 / protein of unknown function (DUF1255) / Joint Center for Structural Genomics / JCSG / Protein Structure Initiative / PSI-2 structural genomics / unknown function / AFE_2634 / protein of unknown function (DUF1255) / Joint Center for Structural Genomics / JCSG / Protein Structure Initiative / PSI-2 | ||||||
| Function / homology | Jelly Rolls / Jelly Rolls / Sandwich / Mainly Beta Function and homology information Function and homology information | ||||||
| Biological species |  Acidithiobacillus ferrooxidans ATCC 23270 (bacteria) Acidithiobacillus ferrooxidans ATCC 23270 (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 0.97 Å | ||||||
 Authors Authors | Joint Center for Structural Genomics (JCSG) | ||||||
 Citation Citation | Journal: To be published Title: Crystal structure of protein of unknown function (DUF1255) (AFE_2634) from ACIDITHIOBACILLUS FERROOXIDANS NCIB8455 at 0.97 A resolution Authors: Joint Center for Structural Genomics (JCSG) | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3eo6.cif.gz | 137.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3eo6.ent.gz | 115.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3eo6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/eo/3eo6ftp://data.pdbj.org/pub/pdb/validation_reports/eo/3eo6 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Refine code: 6 / Auth seq-ID: 0 - 105 / Label seq-ID: 0 - 105
|
-Components
| #1: Protein | Mass: 11677.310 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Acidithiobacillus ferrooxidans ATCC 23270 (bacteria)Gene: AFE_2634 / Plasmid: SpeedET / Production host: Escherichia coli (E. coli) / Strain (production host): HK100#2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-TRS / | Tris  Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 422 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 422 / Source method: isolated from a natural source / Formula: H2OSequence details | (1) THE CONSTRUCT WAS EXPRESSED WITH A PURIFICATION TAG MGSDKIHHHHHHENLYFQG. THE TAG WAS REMOVED ...(1) THE CONSTRUCT WAS EXPRESSED WITH A PURIFICATI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.08 Å3/Da / Density % sol: 40.74 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 0.2000M MgCl2, 30.0000% PEG-4000, 0.1M TRIS pH 8.5, NANODROP, VAPOR DIFFUSION, SITTING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL11-1 / Wavelength: 0.91837,0.97837 / Beamline: BL11-1 / Wavelength: 0.91837,0.97837 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: Aug 1, 2008 / Details: Flat mirror (vertical focusing) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Single crystal Si(111) bent monochromator (horizontal focusing) Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 0.97→28.194 Å / Num. obs: 111566 / % possible obs: 99.4 % / Redundancy: 4.1 % / Biso Wilson estimate: 7.317 Å2 / Rmerge(I) obs: 0.064 / Rsym value: 0.064 / Net I/σ(I): 5.911 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: MAD |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 0.97→28.194 Å / Cor.coef. Fo:Fc: 0.983 / Cor.coef. Fo:Fc free: 0.978 / Occupancy max: 1 / Occupancy min: 0.11 / SU B: 0.613 / SU ML: 0.015 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.022 / ESU R Free: 0.022 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. 2. A MET-INHIBITION PROTOCOL WAS USED FOR SELENOMETHIONINE INCORPORATION DURING PROTEIN EXPRESSION. THE OCCUPANCY OF THE SE ATOMS IN THE ...Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. 2. A MET-INHIBITION PROTOCOL WAS USED FOR SELENOMETHIONINE INCORPORATION DURING PROTEIN EXPRESSION. THE OCCUPANCY OF THE SE ATOMS IN THE MSE RESIDUES WAS REDUCED TO ~0.8 FOR THE REDUCED SCATTERING POWER DUE TO PARTIAL S-MET INCORPORATION. 3. U VALUES WERE REFINED INDIVIDUALLY 4. TRIS (TRS) AND MG MODELED WERE PRESENT IN CRYSTALLIZATION CONDITIONS. 5. THE DENSITY FOR N-TERMINUS AND C-TERMINUS ARE POOR.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 46.85 Å2 / Biso mean: 11.791 Å2 / Biso min: 3.14 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.97→28.194 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Number: 1074 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 0.97→0.995 Å / Total num. of bins used: 20
|
