 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1md2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CHOLERA TOXIN B-PENTAMER WITH DECAVALENT LIGAND BMSC-0013 | ||||||
 Components Components | CHOLERA TOXIN B SUBUNIT | ||||||
 Keywords Keywords | TOXIN / multivalent inhibitor toxin | ||||||
| Function / homology |  Function and homology information Function and homology informationhost cell surface binding / galactose binding / positive regulation of tyrosine phosphorylation of STAT protein / catalytic complex / toxin activity / periplasmic space / host cell plasma membrane / extracellular region / membrane Similarity search - Function | ||||||
| Biological species |  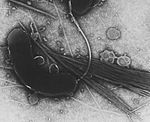 Vibrio cholerae (bacteria) Vibrio cholerae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / isomorphous to 3CHB / Resolution: 1.45 Å X-RAY DIFFRACTION / SYNCHROTRON / isomorphous to 3CHB / Resolution: 1.45 Å | ||||||
 Authors Authors | Zhang, Z. / Merritt, E.A. / Ahn, M. / Roach, C. / Hol, W.G.J. / Fan, E. | ||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2002 Title: Solution and crystallographic studies of branched multivalent ligands that inhibit the receptor-binding of cholera toxin. Authors: Zhang, Z. / Merritt, E.A. / Ahn, M. / Roach, C. / Hou, Z. / Verlinde, C.L. / Hol, W.G. / Fan, E. | ||||||
| History |
| ||||||
| Remark 600 | HETEROGEN Ligand 233 (BMSC-0013) is a decavalent ligand consisting of a pentacyclen core to which ...HETEROGEN Ligand 233 (BMSC-0013) is a decavalent ligand consisting of a pentacyclen core to which are attached five long arms, each containing four 'linker' moieties followed by a squaric acid moiety and then branching to present two monovalent ligands derived from beta-D-galactose. A single copy of the decavalent ligand lies on a 2-fold and straddles two crystallographic asymmetric units. The central core and linker groups do not satisfy crystallographic symmetry and are not visible in the structure and hence not present in this model. The sulfur atom belongs to an unknown species (residue 109, chains D-H) constituting a partial- occupancy improperly formed cysteine disulfide between residues 9 and 86. That is, ~80% of the time there is a 9-86 disulfide and ~20% of the time there is a 9-??? disulfide. | ||||||
| Remark 650 | HELIX DETERMINATION METHOD: AUTHOR PROVIDED | ||||||
| Remark 700 | SHEET DETERMINATION METHOD: AUTHOR PROVIDED |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1md2.cif.gz | 249.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1md2.ent.gz | 204.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1md2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/md/1md2ftp://data.pdbj.org/pub/pdb/validation_reports/md/1md2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3chbS S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | The crystallized complex contains two CTB pentamers sandwiching a single decavalent ligand. This sandwich spans 2 asymmetric units. |
-Components
| #1: Protein | Mass: 11655.332 Da / Num. of mol.: 5 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Vibrio cholerae (bacteria) / Gene: CTXB / Production host: #2: Chemical | ChemComp-233 / [ 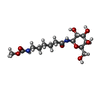  Mass: 350.365 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C14H26N2O8 Mass: 350.365 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C14H26N2O8#3: Chemical | 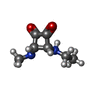  Mass: 156.182 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H12N2O2 / Comment: inhibitor*YM Mass: 156.182 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H12N2O2 / Comment: inhibitor*YM#4: Chemical | ChemComp-CYN / |   Mass: 26.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CN Mass: 26.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CN#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 674 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 674 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.18 Å3/Da / Density % sol: 43.62 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, sitting drop / pH: 7.5 Details: PEG 1000, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature rtK Temp details: room temperature | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-BM / Wavelength: 0.9789 Å / Beamline: 19-BM / Wavelength: 0.9789 Å |
| Detector | Type: CUSTOM-MADE / Detector: CCD / Date: Nov 30, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9789 Å / Relative weight: 1 |
| Reflection | Resolution: 1.4→50 Å / Num. all: 84651 / Num. obs: 84651 / % possible obs: 86 % / Observed criterion σ(I): -3 / Redundancy: 3 % / Biso Wilson estimate: 21 Å2 / Rmerge(I) obs: 0.049 / Net I/σ(I): 18 |
| Reflection shell | Resolution: 1.4→1.45 Å / Redundancy: 1.5 % / Rmerge(I) obs: 0.326 / Mean I/σ(I) obs: 1.4 / Num. unique all: 3176 / % possible all: 32 |
| Reflection | *PLUS Highest resolution: 1.45 Å / Lowest resolution: 50 Å / Num. obs: 81475 / % possible obs: 92 % / Rmerge(I) obs: 0.048 |
| Reflection shell | *PLUS Highest resolution: 1.45 Å / Lowest resolution: 1.51 Å / % possible obs: 54 % / Num. unique obs: 5338 / Rmerge(I) obs: 0.306 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: isomorphous to 3CHB Starting model: PDB entry 3CHB Resolution: 1.45→25 Å / Cor.coef. Fo:Fc: 0.97049 / Cor.coef. Fo:Fc free: 0.95845 / σ(F): 0 / ESU R: 0.07416 / ESU R Free: 0.06325
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→25 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 25 Å / Rfactor Rfree: 0.164 / Rfactor Rwork: 0.125 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: p_bond_d / Dev ideal: 0.018 |
