 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1i7i: CRYSTAL STRUCTURE OF THE LIGAND BINDING DOMAIN OF HUMAN PPAR-GAMM... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1i7i | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE LIGAND BINDING DOMAIN OF HUMAN PPAR-GAMMA IN COMPLEX WITH THE AGONIST AZ 242 | ||||||
 Components Components | PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR GAMMA | ||||||
 Keywords Keywords | TRANSCRIPTION / anti parallel helix sandwich | ||||||
| Function / homology |  Function and homology information Function and homology informationprostaglandin receptor activity / beige fat cell differentiation / negative regulation of receptor signaling pathway via STAT / MECP2 regulates transcription factors / negative regulation of vascular endothelial cell proliferation / negative regulation of extracellular matrix assembly / negative regulation of connective tissue replacement involved in inflammatory response wound healing / positive regulation of cholesterol transport / negative regulation of cellular response to transforming growth factor beta stimulus / arachidonate binding ...prostaglandin receptor activity / beige fat cell differentiation / negative regulation of receptor signaling pathway via STAT / MECP2 regulates transcription factors / negative regulation of vascular endothelial cell proliferation / negative regulation of extracellular matrix assembly / negative regulation of connective tissue replacement involved in inflammatory response wound healing / positive regulation of cholesterol transport / negative regulation of cellular response to transforming growth factor beta stimulus / arachidonate binding / positive regulation of adiponectin secretion / DNA binding domain binding / positive regulation of vascular associated smooth muscle cell apoptotic process / negative regulation of cardiac muscle hypertrophy in response to stress / positive regulation of fatty acid metabolic process / WW domain binding / STAT family protein binding / positive regulation of lipid metabolic process / response to lipid / negative regulation of type II interferon-mediated signaling pathway / LBD domain binding / negative regulation of cholesterol storage / positive regulation of lipoprotein transport / negative regulation of SMAD protein signal transduction / lipid homeostasis / E-box binding / alpha-actinin binding / R-SMAD binding / negative regulation of vascular associated smooth muscle cell proliferation / negative regulation of blood vessel endothelial cell migration / white fat cell differentiation / negative regulation of macrophage derived foam cell differentiation / negative regulation of lipid storage / positive regulation of cholesterol efflux / monocyte differentiation / negative regulation of BMP signaling pathway / cell fate commitment / cellular response to low-density lipoprotein particle stimulus / negative regulation of mitochondrial fission / negative regulation of osteoblast differentiation / long-chain fatty acid transport / positive regulation of fat cell differentiation / BMP signaling pathway / nuclear retinoid X receptor binding / fat cell differentiation / Transcriptional regulation of brown and beige adipocyte differentiation by EBF2 / retinoic acid receptor signaling pathway / negative regulation of MAPK cascade / intracellular receptor signaling pathway / cell maturation / positive regulation of adipose tissue development / hormone-mediated signaling pathway / peroxisome proliferator activated receptor signaling pathway / epithelial cell differentiation / regulation of cellular response to insulin stimulus / peptide binding / response to nutrient / brown fat cell differentiation / negative regulation of miRNA transcription / negative regulation of angiogenesis / placenta development / Regulation of PTEN gene transcription / transcription coregulator binding / positive regulation of apoptotic signaling pathway / SUMOylation of intracellular receptors / negative regulation of smooth muscle cell proliferation / negative regulation of transforming growth factor beta receptor signaling pathway / PPARA activates gene expression / fatty acid metabolic process / regulation of circadian rhythm / Nuclear Receptor transcription pathway / Transcriptional regulation of white adipocyte differentiation / mRNA transcription by RNA polymerase II / positive regulation of miRNA transcription / negative regulation of inflammatory response / DNA-binding transcription repressor activity, RNA polymerase II-specific / regulation of blood pressure / RNA polymerase II transcription regulator complex / nuclear receptor activity / cellular response to insulin stimulus / rhythmic process / glucose homeostasis / MLL4 and MLL3 complexes regulate expression of PPARG target genes in adipogenesis and hepatic steatosis / DNA-binding transcription activator activity, RNA polymerase II-specific / double-stranded DNA binding / cellular response to hypoxia / sequence-specific DNA binding / DNA-binding transcription factor binding / nucleic acid binding / DNA-binding transcription factor activity, RNA polymerase II-specific / cell differentiation / receptor complex / transcription cis-regulatory region binding / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of gene expression / innate immune response / negative regulation of DNA-templated transcription / chromatin binding / positive regulation of gene expression Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.35 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.35 Å | ||||||
 Authors Authors | Petersen, J.F.W. / Cronet, P. / Folmer, R. / Blomberg, N. / Sjoblom, K. / Karlsson, U. / Lindstedt, E.-L. / Bamberg, K. | ||||||
 Citation Citation | Journal: Structure / Year: 2001 Title: Structure of the PPARalpha and -gamma ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Authors: Cronet, P. / Petersen, J.F. / Folmer, R. / Blomberg, N. / Sjoblom, K. / Karlsson, U. / Lindstedt, E.L. / Bamberg, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1i7i.cif.gz | 116.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1i7i.ent.gz | 90.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1i7i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i7/1i7iftp://data.pdbj.org/pub/pdb/validation_reports/i7/1i7i | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1i7gC  2prgS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 33209.383 Da / Num. of mol.: 2 / Fragment: LIGAND BINDING DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  #2: Chemical | 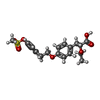  Mass: 408.465 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H24O7S / Comment: agonist*YM Mass: 408.465 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H24O7S / Comment: agonist*YM#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.52 Å3/Da / Density % sol: 51.16 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: sodium citrate, hepes, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Details: Osmic Mirrors |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.35→30 Å / Num. obs: 25345 / % possible obs: 91.3 % / Redundancy: 2.9 % / Biso Wilson estimate: 41.6 Å2 / Rmerge(I) obs: 0.067 / Net I/σ(I): 21.8 |
| Reflection shell | Resolution: 2.35→2.43 Å / Redundancy: 3 % / Rmerge(I) obs: 0.419 / Mean I/σ(I) obs: 2.6 / % possible all: 94.1 |
| Reflection | *PLUS Lowest resolution: 20 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry: 2PRG Resolution: 2.35→19.98 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 1562646.62 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 51.7 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.35→19.98 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.35→2.5 Å / Rfactor Rfree error: 0.024 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 5 % / Rfactor obs: 0.236 / Rfactor Rfree: 0.282 | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 51.5 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.344 / % reflection Rfree: 4.8 % / Rfactor Rwork: 0.324 |
