ジャーナル: Biochemistry / 年: 2008 タイトル: Zinc coordination geometry and ligand binding affinity: the structural and kinetic analysis of the second-shell serine 228 residue and the methionine 180 residue of the aminopeptidase ...タイトル: Zinc coordination geometry and ligand binding affinity: the structural and kinetic analysis of the second-shell serine 228 residue and the methionine 180 residue of the aminopeptidase from Vibrio proteolyticus. 著者: Ataie, N.J. / Hoang, Q.Q. / Zahniser, M.P. / Tu, Y. / Milne, A. / Petsko, G.A. / Ringe, D.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 細菌性ロイシルアミノペプチダーゼ
細菌性ロイシルアミノペプチダーゼ  キーワード
キーワード 機能・相同性情報
機能・相同性情報 Vibrio proteolyticus (バクテリア)
Vibrio proteolyticus (バクテリア) データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード
















 PDBj
PDBj




 集合体
集合体


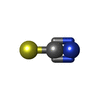


 分子量: 65.409 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Zn
分子量: 65.409 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Zn 分子量: 22.990 Da / 分子数: 8 / 由来タイプ: 合成 / 式: Na
分子量: 22.990 Da / 分子数: 8 / 由来タイプ: 合成 / 式: Na 分子量: 58.082 Da / 分子数: 2 / 由来タイプ: 合成 / 式: CNS
分子量: 58.082 Da / 分子数: 2 / 由来タイプ: 合成 / 式: CNS タイプ: L-peptide linking / 分子量: 131.173 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C6H13NO2
タイプ: L-peptide linking / 分子量: 131.173 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C6H13NO2 試料調製
試料調製 / ビームライン: 14-BM-C / 波長: 0.9001 Å
/ ビームライン: 14-BM-C / 波長: 0.9001 Å 解析
解析