 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9clj | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | HIV-2 CA T=1 Icosahedron; assembled via lipid templating | ||||||||||||||||||||||||
 Components Components | Capsid protein p24 | ||||||||||||||||||||||||
 Keywords Keywords | VIRAL PROTEIN / HIV-2 / Capsid / IP6 | ||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationHIV-2 retropepsin / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / viral penetration into host nucleus ...HIV-2 retropepsin / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / viral penetration into host nucleus / host multivesicular body / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / host cell / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / symbiont-mediated suppression of host gene expression / viral translational frameshifting / symbiont entry into host cell / lipid binding / host cell plasma membrane / host cell nucleus / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||||||||||||||||||||
| Biological species |  Human immunodeficiency virus 2 Human immunodeficiency virus 2 | ||||||||||||||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 1.98 Å | ||||||||||||||||||||||||
 Authors Authors | Cook, M. / Freniere, C. / Xiong, Y. | ||||||||||||||||||||||||
| Funding support |  United States, 3items United States, 3items
| ||||||||||||||||||||||||
 Citation Citation | Journal: Cell Rep / Year: 2025 Title: Structural insights into HIV-2 CA lattice formation and FG-pocket binding revealed by single-particle cryo-EM. Authors: Matthew Cook / Christian Freniere / Chunxiang Wu / Faith Lozano / Yong Xiong / Abstract: One of the striking features of human immunodeficiency virus (HIV) is the capsid, a fullerene cone comprised of pleomorphic capsid protein (CA) that shields the viral genome and recruits cofactors. ...One of the striking features of human immunodeficiency virus (HIV) is the capsid, a fullerene cone comprised of pleomorphic capsid protein (CA) that shields the viral genome and recruits cofactors. Despite significant advances in understanding the mechanisms of HIV-1 CA assembly and host factor interactions, HIV-2 CA assembly remains poorly understood. By templating the assembly of HIV-2 CA on functionalized liposomes, we report high-resolution structures of the HIV-2 CA lattice, including both CA hexamers and pentamers, alone and with peptides of host phenylalanine-glycine (FG)-motif proteins Nup153 and CPSF6. While the overall fold and mode of FG-peptide binding is conserved with HIV-1, this study reveals distinctive features of the HIV-2 CA lattice, including differing structural character at regions of host factor interactions and divergence in the mechanism of formation of CA hexamers and pentamers. This study extends our understanding of HIV capsids and highlights an approach facilitating the study of lentiviral capsid biology. | ||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9clj.cif.gz | 58.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9clj.ent.gz | 39.5 KB | Display | PDB format |
| PDBx/mmJSON format | 9clj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cl/9cljftp://data.pdbj.org/pub/pdb/validation_reports/cl/9clj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  45676MC  9cnsC  9cntC  9cnuC  9cnvC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 | x 60
|
| 2 |
|
| 3 | x 5
|
| 4 | x 6
|
| 5 | 
|
| Symmetry | Point symmetry: (Schoenflies symbol: I (icosahedral)) |
-Components
| #1: Protein | Mass: 26809.490 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: Full length HIV-2 GL-AN capsid protein with a C-terminal Gly-Ser-Ser linker to a hexahistidine tag following proteolytic processing of N-terminal Met. Source: (gene. exp.) Human immunodeficiency virus 2 / Strain: GL-AN / Gene: gag-pol / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | 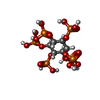  Mass: 660.035 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H18O24P6 Mass: 660.035 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H18O24P6#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | Has protein modification | N | |
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: HIV-2 capsid protein assembled into a lattice via lipid templating. Type: COMPLEX Details: C-terminally hexahistidine tagged HIV-2 CA associated with a micelle decorated with NiNTA headgroups which results in the assembly of an icosahedral lattice of CA. Entity ID: #1 / Source: RECOMBINANT | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 26.9 kDa/nm / Experimental value: NO | |||||||||||||||||||||||||
| Source (natural) | Organism: Human immunodeficiency virus 2 / Strain: GL-AN | |||||||||||||||||||||||||
| Source (recombinant) | Organism: | |||||||||||||||||||||||||
| Buffer solution | pH: 7 Details: The mixed buffer of storage buffer for the protein and lipid components with IP6 supplemented. | |||||||||||||||||||||||||
| Buffer component |
| |||||||||||||||||||||||||
| Specimen | Conc.: 10.7 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES Details: Sample was prepared with 400 uM HIV-2 CA-6xHis protein, 5.9 mM lipid mix (described in publication), and 4 mM IP6. Sample was well-distributed on the grid, mostly monodisperse. Perhaps ...Details: Sample was prepared with 400 uM HIV-2 CA-6xHis protein, 5.9 mM lipid mix (described in publication), and 4 mM IP6. Sample was well-distributed on the grid, mostly monodisperse. Perhaps slightly more particles on carbon versus in the hole. | |||||||||||||||||||||||||
| Specimen support | Details: 15 mA discharge current. / Grid material: COPPER / Grid mesh size: 200 divisions/in. / Grid type: Quantifoil R2/1 | |||||||||||||||||||||||||
| Vitrification | Instrument: FEI VITROBOT MARK IV / Cryogen name: ETHANE / Humidity: 100 % / Chamber temperature: 298 K Details: Grids were dual-side blotted with blot force 0 for 5.5 sec before plunge freezing in liquid ethane. |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 81000 X / Nominal defocus max: 2000 nm / Nominal defocus min: 800 nm / Cs: 2.7 mm / C2 aperture diameter: 30 µm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Image recording | Electron dose: 50 e/Å2 / Film or detector model: GATAN K3 (6k x 4k) |
| EM imaging optics | Energyfilter name: GIF Quantum LS / Energyfilter slit width: 15 eV |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 690629 | ||||||||||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: I (icosahedral) | ||||||||||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 1.98 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 74821 / Symmetry type: POINT | ||||||||||||||||||||||||||||||||||||||||
| Atomic model building | Protocol: FLEXIBLE FIT / Space: REAL / Target criteria: Cross-correlation coefficient Details: An HIV-1 CA chain from pentamers (PDB: 8CKW) was initially fit into the density by rigid body fitting. It was then mutated to match the HIV-2 sequence, with unmodeled residues being replaced ...Details: An HIV-1 CA chain from pentamers (PDB: 8CKW) was initially fit into the density by rigid body fitting. It was then mutated to match the HIV-2 sequence, with unmodeled residues being replaced before flexible fitting and refinement into the map volume. | ||||||||||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 8CKW Pdb chain-ID: A / Accession code: 8CKW / Chain residue range: 1-221 Details: Due to structural similarities expected between HIV-2 and HIV-1, an HIV-1 model was used for initial fitting. Pdb chain residue range: 1-221 / Source name: PDB / Type: experimental model |

