 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8v3n | ||||||
|---|---|---|---|---|---|---|---|
| Title | CCP5 in complex with Glu-P-Glu transition state analog | ||||||
 Components Components | Cytosolic carboxypeptidase-like protein 5 | ||||||
 Keywords Keywords | HYDROLASE/INHIBITOR / carboxypeptidase deglutamylation branch glutamate removal microtubule / HYDROLASE / HYDROLASE-INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationtubulin-glutamate carboxypeptidase / protein deglutamylation / C-terminal protein deglutamylation / protein side chain deglutamylation / protein branching point deglutamylation / Carboxyterminal post-translational modifications of tubulin / Hydrolases; Acting on peptide bonds (peptidases); Metallocarboxypeptidases / metallocarboxypeptidase activity / tubulin binding / mitotic spindle ...tubulin-glutamate carboxypeptidase / protein deglutamylation / C-terminal protein deglutamylation / protein side chain deglutamylation / protein branching point deglutamylation / Carboxyterminal post-translational modifications of tubulin / Hydrolases; Acting on peptide bonds (peptidases); Metallocarboxypeptidases / metallocarboxypeptidase activity / tubulin binding / mitotic spindle / microtubule cytoskeleton / midbody / defense response to virus / proteolysis / zinc ion binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Chen, J. / Zehr, E.A. / Gruschus, J.M. / Szyk, A. / Liu, Y. / Tanner, M.E. / Tjandra, N. / Roll-Mecak, A. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Nature / Year: 2024 Title: Tubulin code eraser CCP5 binds branch glutamates by substrate deformation. Authors: Jiayi Chen / Elena A Zehr / James M Gruschus / Agnieszka Szyk / Yanjie Liu / Martin E Tanner / Nico Tjandra / Antonina Roll-Mecak /  Abstract: Microtubule function is modulated by the tubulin code, diverse posttranslational modifications that are altered dynamically by writer and eraser enzymes. Glutamylation-the addition of branched ...Microtubule function is modulated by the tubulin code, diverse posttranslational modifications that are altered dynamically by writer and eraser enzymes. Glutamylation-the addition of branched (isopeptide-linked) glutamate chains-is the most evolutionarily widespread tubulin modification. It is introduced by tubulin tyrosine ligase-like enzymes and erased by carboxypeptidases of the cytosolic carboxypeptidase (CCP) family. Glutamylation homeostasis, achieved through the balance of writers and erasers, is critical for normal cell function, and mutations in CCPs lead to human disease. Here we report cryo-electron microscopy structures of the glutamylation eraser CCP5 in complex with the microtubule, and X-ray structures in complex with transition-state analogues. Combined with NMR analysis, these analyses show that CCP5 deforms the tubulin main chain into a unique turn that enables lock-and-key recognition of the branch glutamate in a cationic pocket that is unique to CCP family proteins. CCP5 binding of the sequences flanking the branch point primarily through peptide backbone atoms enables processing of diverse tubulin isotypes and non-tubulin substrates. Unexpectedly, CCP5 exhibits inefficient processing of an abundant β-tubulin isotype in the brain. This work provides an atomistic view into glutamate branch recognition and resolution, and sheds light on homeostasis of the tubulin glutamylation syntax. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8v3n.cif.gz | 215.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8v3n.ent.gz | Display | PDB format | |
| PDBx/mmJSON format | 8v3n.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v3/8v3nftp://data.pdbj.org/pub/pdb/validation_reports/v3/8v3n | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  8v3mC  8v3oC  8v3pC  8v3qC  8v3rC  8v3sC  8v4kC  8v4lC  8v4mC C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 59170.668 Da / Num. of mol.: 1 / Mutation: E516A,residues 339-424 replaced with a SGSGG loop Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: AGBL5, CCP5 / Plasmid: pFastBac / Details (production host): His_MBP_Asn10_TEV / Cell line (production host): Sf9 / Production host:   Spodoptera frugiperda (fall armyworm) / Strain (production host): IPLB-Sf-21-AE Spodoptera frugiperda (fall armyworm) / Strain (production host): IPLB-Sf-21-AEReferences: UniProt: Q8NDL9, Hydrolases; Acting on peptide bonds (peptidases); Metallocarboxypeptidases, tubulin-glutamate carboxypeptidase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||||||
| #3: Chemical | 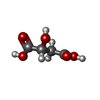  Mass: 134.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O5 Mass: 134.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O5#4: Chemical | ChemComp-A1AAG / ( | Mass: 380.331 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H25N2O8P / Feature type: SUBJECT OF INVESTIGATION #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 170 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 170 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 46.14 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop Details: 0.15 M DL-Malic acid, pH7.0, 0.1 M imidazole, pH7.0, 16% PEG MME 550 |
-Data collection
| Diffraction | Mean temperature: 93 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 24-ID-E / Wavelength: 0.9792 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Feb 10, 2022 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9792 Å / Relative weight: 1 |
| Reflection | Resolution: 2.27→103.23 Å / Num. obs: 25523 / % possible obs: 99.4 % / Redundancy: 12.9 % / CC1/2: 0.961 / Rmerge(I) obs: 0.306 / Rrim(I) all: 0.334 / Net I/σ(I): 11.1 |
| Reflection shell | Resolution: 2.27→2.35 Å / Redundancy: 12.5 % / Rmerge(I) obs: 1.869 / Mean I/σ(I) obs: 2.2 / Num. unique obs: 2238 / CC1/2: 0.613 / Rrim(I) all: 1.992 / % possible all: 95.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.3→65.11 Å / SU ML: 0.2 / Cross valid method: FREE R-VALUE / σ(F): 1.33 / Phase error: 22.87 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→65.11 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
