 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7p2l: thermostabilised 7TM domain of human mGlu5 receptor bound to phot... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7p2l | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | thermostabilised 7TM domain of human mGlu5 receptor bound to photoswitchable ligand alloswitch-1 | |||||||||
 Components Components | Metabotropic glutamate receptor 5,Endolysin,Metabotropic glutamate receptor 5 | |||||||||
 Keywords Keywords | SIGNALING PROTEIN / G protein coupled receptor / mGlu5 7TM / photoswitchable ligand / allosteric modulator | |||||||||
| Function / homology |  Function and homology information Function and homology informationA2A adenosine receptor binding / sensory perception of hot stimulus / negative regulation of dendritic spine morphogenesis / : / G protein-coupled receptor activity involved in regulation of postsynaptic membrane potential / adenylate cyclase inhibiting G protein-coupled glutamate receptor activity / protein localization to nuclear inner membrane / positive regulation of cellular response to hypoxia / phospholipase C-activating G protein-coupled glutamate receptor signaling pathway / operant conditioning ...A2A adenosine receptor binding / sensory perception of hot stimulus / negative regulation of dendritic spine morphogenesis / : / G protein-coupled receptor activity involved in regulation of postsynaptic membrane potential / adenylate cyclase inhibiting G protein-coupled glutamate receptor activity / protein localization to nuclear inner membrane / positive regulation of cellular response to hypoxia / phospholipase C-activating G protein-coupled glutamate receptor signaling pathway / operant conditioning / positive regulation of long-term neuronal synaptic plasticity / positive regulation of sensory perception of pain / negative regulation of excitatory postsynaptic potential / positive regulation of dopamine secretion / desensitization of G protein-coupled receptor signaling pathway / G protein-coupled glutamate receptor signaling pathway / positive regulation of neural precursor cell proliferation / Class C/3 (Metabotropic glutamate/pheromone receptors) / nuclear inner membrane / glutamate receptor activity / Neurexins and neuroligins / astrocyte projection / response to corticosterone / response to morphine / temperature homeostasis / protein tyrosine kinase activator activity / viral release from host cell by cytolysis / conditioned place preference / regulation of synaptic transmission, glutamatergic / peptidoglycan catabolic process / regulation of long-term synaptic depression / positive regulation of calcium-mediated signaling / protein tyrosine kinase binding / dendritic shaft / response to amphetamine / locomotory behavior / synapse organization / postsynaptic density membrane / Schaffer collateral - CA1 synapse / cognition / G protein-coupled receptor activity / cellular response to amyloid-beta / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / positive regulation of cytosolic calcium ion concentration / dendritic spine / G alpha (q) signalling events / host cell cytoplasm / chemical synaptic transmission / response to ethanol / learning or memory / positive regulation of MAPK cascade / defense response to bacterium / response to antibiotic / neuronal cell body / regulation of DNA-templated transcription / dendrite / glutamatergic synapse / identical protein binding / plasma membrane / cytoplasm Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) Enterobacteria phage T4 (virus) Enterobacteria phage T4 (virus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.54 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.54 Å | |||||||||
 Authors Authors | Huang, C.Y. / Vinothkumar, K.R. / Lebon, G. | |||||||||
| Funding support |  France, 2items France, 2items
| |||||||||
 Citation Citation | Journal: Cell Rep / Year: 2021 Title: Agonists and allosteric modulators promote signaling from different metabotropic glutamate receptor 5 conformations. Authors: Chady Nasrallah / Giuseppe Cannone / Julie Briot / Karine Rottier / Alice E Berizzi / Chia-Ying Huang / Robert B Quast / Francois Hoh / Jean-Louis Banères / Fanny Malhaire / Ludovic Berto / ...Authors: Chady Nasrallah / Giuseppe Cannone / Julie Briot / Karine Rottier / Alice E Berizzi / Chia-Ying Huang / Robert B Quast / Francois Hoh / Jean-Louis Banères / Fanny Malhaire / Ludovic Berto / Anaëlle Dumazer / Joan Font-Ingles / Xavier Gómez-Santacana / Juanlo Catena / Julie Kniazeff / Cyril Goudet / Amadeu Llebaria / Jean-Philippe Pin / Kutti R Vinothkumar / Guillaume Lebon /     Abstract: Metabotropic glutamate receptors (mGluRs) are dimeric G-protein-coupled receptors activated by the main excitatory neurotransmitter, L-glutamate. mGluR activation by agonists binding in the venus ...Metabotropic glutamate receptors (mGluRs) are dimeric G-protein-coupled receptors activated by the main excitatory neurotransmitter, L-glutamate. mGluR activation by agonists binding in the venus flytrap domain is regulated by positive (PAM) or negative (NAM) allosteric modulators binding to the 7-transmembrane domain (7TM). We report the cryo-electron microscopy structures of fully inactive and intermediate-active conformations of mGlu receptor bound to an antagonist and a NAM or an agonist and a PAM, respectively, as well as the crystal structure of the 7TM bound to a photoswitchable NAM. The agonist induces a large movement between the subunits, bringing the 7TMs together and stabilizing a 7TM conformation structurally similar to the inactive state. Using functional approaches, we demonstrate that the PAM stabilizes a 7TM active conformation independent of the conformational changes induced by agonists, representing an alternative mode of mGlu activation. These findings provide a structural basis for different mGluR activation modes. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7p2l.cif.gz | 99.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7p2l.ent.gz | 72.2 KB | Display | PDB format |
| PDBx/mmJSON format | 7p2l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p2/7p2lftp://data.pdbj.org/pub/pdb/validation_reports/p2/7p2l | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7fd8C  7fd9C  4oo9S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 51918.055 Da / Num. of mol.: 1 / Mutation: E579A,N667Y,I669A,G675M,T742A,S753A Source method: isolated from a genetically manipulated source Details: The macromolecule is a chimera of mGluR TMD and T4 lysozyme. The Uniprot ID of mGluR5 is P41594 and the T4 lysozyme if P00720. The lysozyme sequence is inserted between residue 678 and 679 ...Details: The macromolecule is a chimera of mGluR TMD and T4 lysozyme. The Uniprot ID of mGluR5 is P41594 and the T4 lysozyme if P00720. The lysozyme sequence is inserted between residue 678 and 679 of mGluR5. In the PDB, the lysozyme is numbered from 1002-1162 (Asn & Tyr). The gaps are between 680-688 and 721-728. Residues 644 & 733 form a disuphide bond. The lysozyme has also been engineered and has the following mutations. C54T (in PDB it is 1054) and C97T (in PDB it is 1097). Source: (gene. exp.) Homo sapiens (human), (gene. exp.) Enterobacteria phage T4 (virus)Gene: GRM5, GPRC1E, MGLUR5 / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P41594, UniProt: P00720, lysozyme Spodoptera frugiperda (fall armyworm) / References: UniProt: P41594, UniProt: P00720, lysozyme |
|---|---|
| #2: Chemical | ChemComp-4YI / 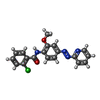  Mass: 366.801 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H15ClN4O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 366.801 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H15ClN4O2 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.72 Å3/Da / Density % sol: 54.83 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: lipidic cubic phase Details: 0.15-0.25 M ammonium phosphate dibasic, 22-24 % polyethylene glycol 400, either with 0.10 M 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.7-6.8 or 0.1 M HEPES pH 6.8 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS / Beamline: X06SA / Wavelength: 1 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Dec 14, 2020 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.54→49.11 Å / Num. obs: 16595 / % possible obs: 100 % / Redundancy: 10.66 % / CC1/2: 0.997 / Rrim(I) all: 0.253 / Net I/σ(I): 6.35 |
| Reflection shell | Resolution: 2.54→2.61 Å / Redundancy: 9.3 % / Num. unique obs: 1184 / CC1/2: 0.51 / Rrim(I) all: 2.685 / % possible all: 99 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4oo9 Resolution: 2.54→49.11 Å / Cor.coef. Fo:Fc: 0.937 / Cor.coef. Fo:Fc free: 0.881 / SU B: 17.497 / SU ML: 0.35 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.647 / ESU R Free: 0.336 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 149.46 Å2 / Biso mean: 69.807 Å2 / Biso min: 46.68 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.54→49.11 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.54→2.606 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
