 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7nqg: The structure of the SBP TarP_Rhp in complex with 4-hydroxyphenyl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7nqg | ||||||
|---|---|---|---|---|---|---|---|
| Title | The structure of the SBP TarP_Rhp in complex with 4-hydroxyphenylacetate | ||||||
 Components Components | TrapT family, dctP subunit, C4-dicarboxylate periplasmic binding protein | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / periplasmic binding protein / TRAP transporter / solute binding protein / hydroxycinnamate | ||||||
| Function / homology | TRAP transporter solute receptor DctP / TRAP transporter solute receptor DctP superfamily / Bacterial extracellular solute-binding protein, family 7 / transmembrane transport / 4-HYDROXYPHENYLACETATE / PHOSPHATE ION / TRAP transporter substrate-binding protein Function and homology information Function and homology information | ||||||
| Biological species |  Rhodopseudomonas palustris (phototrophic) Rhodopseudomonas palustris (phototrophic) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO PHASING / Resolution: 1.1 Å X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO PHASING / Resolution: 1.1 Å | ||||||
 Authors Authors | Bisson, C. / Salmon, R.C. / West, L. / Rafferty, J.B. / Hitchcock, A. / Thomas, G.H. / Kelly, D.J. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: Febs J. / Year: 2022 Title: The structural basis for high-affinity uptake of lignin-derived aromatic compounds by proteobacterial TRAP transporters. Authors: Bisson, C. / Salmon, R.C. / West, L. / Rafferty, J.B. / Hitchcock, A. / Thomas, G.H. / Kelly, D.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7nqg.cif.gz | 145.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7nqg.ent.gz | Display | PDB format | |
| PDBx/mmJSON format | 7nqg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nq/7nqgftp://data.pdbj.org/pub/pdb/validation_reports/nq/7nqg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7nr2C  7nraC  7nrrC  7nswC  7ntdC  7nteC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 36450.441 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: M1-D4 sequence not visible in structure. N-terminal residue was found to be a PCA, formed by spontaneous cyclisation on an N-terminal glutamine (Q5). 6xHis tag on C-terminus. Source: (gene. exp.) Rhodopseudomonas palustris (strain ATCC BAA-98 / CGA009) (phototrophic)Strain: ATCC BAA-98 / CGA009 / Gene: RPA1782 / Plasmid: pET22b+ (pelB) / Production host: | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-4HP / 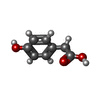  Mass: 152.147 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H8O3 Mass: 152.147 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H8O3 | ||||||||
| #3: Chemical |   Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical | ChemComp-PO4 / |   Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 373 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 373 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.98 Å3/Da / Density % sol: 37.84 % / Description: Plates |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, sitting drop / pH: 8 Details: 0.1 M Tris-HCl pH 8 and 20 % (w/v) PEG 6000 (+6 mM coumarate - not bound) |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I03 / Wavelength: 0.97625 Å / Beamline: I03 / Wavelength: 0.97625 Å |
| Detector | Type: DECTRIS EIGER X 9M / Detector: PIXEL / Date: May 29, 2014 |
| Radiation | Monochromator: Synchrotron / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97625 Å / Relative weight: 1 |
| Reflection | Resolution: 1.1→50.5 Å / Num. obs: 113835 / % possible obs: 99.6 % / Redundancy: 6.3 % / Rmerge(I) obs: 0.05 / Rpim(I) all: 0.023 / Net I/σ(I): 17 |
| Reflection shell | Resolution: 1.1→1.13 Å / Redundancy: 3 % / Rmerge(I) obs: 0.436 / Mean I/σ(I) obs: 2.2 / Num. unique obs: 7978 / Rpim(I) all: 0.328 / % possible all: 96.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: AB INITIO PHASING / Resolution: 1.1→47.647 Å / Cor.coef. Fo:Fc: 0.982 / Cor.coef. Fo:Fc free: 0.978 / SU B: 0.767 / SU ML: 0.016 / Cross valid method: FREE R-VALUE / ESU R: 0.025 / ESU R Free: 0.026 Details: Hydrogens have been added in their riding positions
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 11.509 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.1→47.647 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
