 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7egl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Bicarbonate transporter complex SbtA-SbtB bound to HCO3- | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSPORT PROTEIN / Bicarbonate transporter / CCM / allosteric inhibition / photosynthesis | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of nitrogen utilization / plasma membrane-derived thylakoid membrane / enzyme regulator activity Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.2 Å | ||||||
 Authors Authors | Fang, S. / Huang, X. / Zhang, X. / Zhang, P. | ||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2021 Title: Molecular mechanism underlying transport and allosteric inhibition of bicarbonate transporter SbtA. Authors: Sunzhenhe Fang / Xiaowei Huang / Xue Zhang / Minhua Zhang / Yahui Hao / Hui Guo / Lu-Ning Liu / Fang Yu / Peng Zhang /   Abstract: SbtA is a high-affinity, sodium-dependent bicarbonate transporter found in the cyanobacterial CO-concentrating mechanism (CCM). SbtA forms a complex with SbtB, while SbtB allosterically regulates the ...SbtA is a high-affinity, sodium-dependent bicarbonate transporter found in the cyanobacterial CO-concentrating mechanism (CCM). SbtA forms a complex with SbtB, while SbtB allosterically regulates the transport activity of SbtA by binding with adenyl nucleotides. The underlying mechanism of transport and regulation of SbtA is largely unknown. In this study, we report the three-dimensional structures of the cyanobacterial sp. PCC 6803 SbtA-SbtB complex in both the presence and absence of HCO and/or AMP at 2.7 Å and 3.2 Å resolution. An analysis of the inward-facing state of the SbtA structure reveals the HCO/Na binding site, providing evidence for the functional unit as a trimer. A structural comparison found that SbtA adopts an elevator mechanism for bicarbonate transport. A structure-based analysis revealed that the allosteric inhibition of SbtA by SbtB occurs mainly through the T-loop of SbtB, which binds to both the core domain and the scaffold domain of SbtA and locks it in an inward-facing state. T-loop conformation is stabilized by the AMP molecules binding at the SbtB trimer interfaces and may be adjusted by other adenyl nucleotides. The unique regulatory mechanism of SbtA by SbtB makes it important to study inorganic carbon uptake systems in CCM, which can be used to modify photosynthesis in crops. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7egl.cif.gz | 173.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7egl.ent.gz | 137.1 KB | Display | PDB format |
| PDBx/mmJSON format | 7egl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/eg/7eglftp://data.pdbj.org/pub/pdb/validation_reports/eg/7egl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7egkSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39671.246 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: slr1512 / Production host: |
|---|---|
| #2: Protein | Mass: 12021.810 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: slr1513 / Production host: |
| #3: Chemical | ChemComp-BCT / 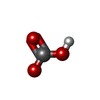  Mass: 61.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CHO3 / Feature type: SUBJECT OF INVESTIGATION / Comment: pH buffer*YM Mass: 61.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CHO3 / Feature type: SUBJECT OF INVESTIGATION / Comment: pH buffer*YM |
| #4: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na / Feature type: SUBJECT OF INVESTIGATION Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na / Feature type: SUBJECT OF INVESTIGATION |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.77 Å3/Da / Density % sol: 67.37 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: vapor diffusion, sitting drop / pH: 9 Details: 0.05M magnesium chloride, 0.1M glycine, 22% PEG 400 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL19U1 / Wavelength: 0.9798 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jun 26, 2020 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9798 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 3.2→50 Å / Num. obs: 15727 / % possible obs: 100 % / Redundancy: 9.6 % / Rmerge(I) obs: 0.162 / Rpim(I) all: 0.054 / Rrim(I) all: 0.171 / Χ2: 0.966 / Net I/σ(I): 3.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / % possible all: 100
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 7EGK Resolution: 3.2→48.774 Å / SU ML: 0.43 / Cross valid method: THROUGHOUT / σ(F): 1.35 / Phase error: 32.95 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 111.48 Å2 / Biso mean: 40.1048 Å2 / Biso min: 11.62 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 3.2→48.774 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -11.4841 Å / Origin y: 44.1124 Å / Origin z: 25.149 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
