 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7e4y | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of tubulin in complex with L-DM4-SMe | ||||||
 Components Components |
| ||||||
 Keywords Keywords | STRUCTURAL PROTEIN/INHIBITOR / microtubule assembly inhibitors / STRUCTURAL PROTEIN-INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationtubulin-tyrosine ligase activity / positive regulation of axon guidance / microtubule depolymerization / regulation of microtubule polymerization or depolymerization / microtubule-based process / cytoplasmic microtubule / cellular response to interleukin-4 / protein modification process / spindle microtubule / tubulin binding ...tubulin-tyrosine ligase activity / positive regulation of axon guidance / microtubule depolymerization / regulation of microtubule polymerization or depolymerization / microtubule-based process / cytoplasmic microtubule / cellular response to interleukin-4 / protein modification process / spindle microtubule / tubulin binding / structural constituent of cytoskeleton / microtubule cytoskeleton organization / neuron migration / neuron projection development / mitotic cell cycle / double-stranded RNA binding / growth cone / microtubule cytoskeleton / microtubule / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / neuron projection / cilium / protein heterodimerization activity / nucleotide binding / GTPase activity / ubiquitin protein ligase binding / GTP binding / Golgi apparatus / metal ion binding / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |   | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.708 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.708 Å | ||||||
 Authors Authors | Wang, Y. / Li, W. | ||||||
| Funding support |  China, 1items China, 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: C3 Ester Side Chain Plays a Pivotal Role in the Antitumor Activity of Maytansinoids Authors: Wang, Y. / Li, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7e4y.cif.gz | 818.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7e4y.ent.gz | 675.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7e4y.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e4/7e4yftp://data.pdbj.org/pub/pdb/validation_reports/e4/7e4y | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7e4pC  4tv8S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 4 types, 6 molecules ACBDEF
| #1: Protein | Mass: 48966.324 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Protein | Mass: 48391.410 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #3: Protein | | Mass: 16282.527 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #4: Protein | | Mass: 43825.914 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|
-Non-polymers , 8 types, 154 molecules 




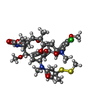
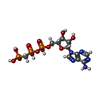

| #5: Chemical |  Mass: 523.180 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM Mass: 523.180 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM#6: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Mg#7: Chemical |  Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#8: Chemical | ChemComp-GDP / |  Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM#9: Chemical | ChemComp-MES / |  Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM#10: Chemical | ChemComp-BKU / ( |  Mass: 812.432 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C38H54ClN3O10S2 / Feature type: SUBJECT OF INVESTIGATION Mass: 812.432 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C38H54ClN3O10S2 / Feature type: SUBJECT OF INVESTIGATION#11: Chemical | ChemComp-ACP / |  Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM#12: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 140 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.83 Å3/Da / Density % sol: 56.48 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.7 Details: 6% poly(ethylene glycol) 4000, 8% glycerol, 0.1 M MES (pH 6.7), 30 mM CaCl2, 30 mM MgCl2 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL19U1 / Wavelength: 0.9798 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jul 4, 2018 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: M / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9798 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.7→40 Å / Num. obs: 81337 / % possible obs: 100 % / Redundancy: 13.2 % / Biso Wilson estimate: 55.61 Å2 / Rmerge(I) obs: 0.115 / Rpim(I) all: 0.033 / Rrim(I) all: 0.12 / Χ2: 0.685 / Net I/σ(I): 4.4 / Num. measured all: 1071851 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / % possible all: 100
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4TV8 Resolution: 2.708→39.512 Å / SU ML: 0.34 / Cross valid method: THROUGHOUT / σ(F): 1.36 / Phase error: 24.96 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 295.34 Å2 / Biso mean: 61.08 Å2 / Biso min: 17.32 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.708→39.512 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
|
