| 登録情報 | データベース: PDB / ID: 7da6
|
|---|
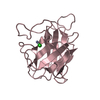
| タイトル | Enterovirus 71 2A Protease mutant- C110A in complex with peptide inhibitor |
|---|
 要素 要素 | - PHE-ARG-GLY-LYS
- Polyprotein
|
|---|
 キーワード キーワード | VIRAL PROTEIN / 2A protease |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
viral process / peptidase activity / host cell cytoplasm / proteolysis / cytoplasm類似検索 - 分子機能 Peptidase C3, picornavirus core protein 2A / Picornavirus core protein 2A / Peptidase S1, PA clan, chymotrypsin-like fold / Peptidase S1, PA clan類似検索 - ドメイン・相同性 |
|---|
| 生物種 |   Human enterovirus 71 (エンテロウイルス) Human enterovirus 71 (エンテロウイルス)
synthetic construct (人工物) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 2.2 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 2.2 Å |
|---|
 データ登録者 データ登録者 | Yang, W.Z. / Yuan, H.S. |
|---|
| 資金援助 | 1件 |
|---|
 引用 引用 | ジャーナル: Acs Bio Med Chem Au / 年: 2022
タイトル: Efficient Strategy to Design Protease Inhibitors: Application to Enterovirus 71 2A Protease.
著者: Chen, T. / Grauffel, C. / Yang, W.Z. / Chen, Y.P. / Yuan, H.S. / Lim, C. |
|---|
| 履歴 | | 登録 | 2020年10月15日 | 登録サイト: PDBJ / 処理サイト: PDBJ |
|---|
| 改定 1.0 | 2021年8月25日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 2.0 | 2021年11月10日 | Group: Advisory / Atomic model ...Advisory / Atomic model / Author supporting evidence / Data collection / Database references / Derived calculations / Experimental preparation / Polymer sequence / Refinement description / Source and taxonomy / Structure summary
カテゴリ: atom_site / entity ...atom_site / entity / entity_poly / entity_poly_seq / entity_src_gen / exptl_crystal / pdbx_audit_support / pdbx_contact_author / pdbx_entity_src_syn / pdbx_nonpoly_scheme / pdbx_poly_seq_scheme / pdbx_struct_conn_angle / pdbx_struct_sheet_hbond / pdbx_unobs_or_zero_occ_atoms / pdbx_unobs_or_zero_occ_residues / pdbx_validate_close_contact / pdbx_validate_rmsd_angle / pdbx_validate_rmsd_bond / pdbx_validate_symm_contact / pdbx_validate_torsion / refine / refine_hist / refine_ls_restr / refine_ls_shell / reflns / reflns_shell / software / struct / struct_conf / struct_conn / struct_ref / struct_ref_seq / struct_ref_seq_dif / struct_sheet_range
Item: _entity.formula_weight / _entity.pdbx_description ..._entity.formula_weight / _entity.pdbx_description / _entity_poly.pdbx_seq_one_letter_code / _entity_poly.pdbx_seq_one_letter_code_can / _entity_src_gen.gene_src_common_name / _entity_src_gen.pdbx_end_seq_num / _exptl_crystal.density_Matthews / _exptl_crystal.density_percent_sol / _pdbx_entity_src_syn.pdbx_end_seq_num / _pdbx_nonpoly_scheme.auth_seq_num / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _pdbx_struct_sheet_hbond.range_1_auth_comp_id / _pdbx_struct_sheet_hbond.range_1_auth_seq_id / _pdbx_struct_sheet_hbond.range_1_label_comp_id / _pdbx_struct_sheet_hbond.range_1_label_seq_id / _pdbx_struct_sheet_hbond.range_2_auth_comp_id / _pdbx_struct_sheet_hbond.range_2_auth_seq_id / _pdbx_struct_sheet_hbond.range_2_label_comp_id / _pdbx_struct_sheet_hbond.range_2_label_seq_id / _pdbx_validate_rmsd_angle.angle_deviation / _pdbx_validate_rmsd_angle.angle_value / _refine.B_iso_max / _refine.B_iso_mean / _refine.B_iso_min / _refine.ls_R_factor_R_free / _refine.ls_R_factor_R_work / _refine.ls_R_factor_obs / _refine.ls_d_res_high / _refine.ls_number_reflns_R_work / _refine.ls_number_reflns_obs / _refine.ls_percent_reflns_obs / _refine.pdbx_overall_phase_error / _refine_hist.d_res_high / _refine_hist.number_atoms_total / _refine_hist.pdbx_B_iso_mean_ligand / _refine_hist.pdbx_B_iso_mean_solvent / _refine_hist.pdbx_number_atoms_protein / _refine_ls_restr.dev_ideal / _refine_ls_restr.number / _reflns.B_iso_Wilson_estimate / _reflns.d_resolution_high / _reflns.d_resolution_low / _reflns.pdbx_CC_half / _reflns.pdbx_Rpim_I_all / _reflns.pdbx_Rrim_I_all / _reflns.pdbx_chi_squared / _reflns.pdbx_netI_over_sigmaI / _reflns.pdbx_number_measured_all / _software.classification / _software.name / _software.version / _struct.title / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_label_seq_id / _struct_ref.db_code / _struct_ref.pdbx_align_begin / _struct_ref.pdbx_db_accession / _struct_ref.pdbx_seq_one_letter_code / _struct_ref_seq.db_align_beg / _struct_ref_seq.db_align_end / _struct_ref_seq.pdbx_auth_seq_align_beg / _struct_ref_seq.pdbx_auth_seq_align_end / _struct_ref_seq.pdbx_db_accession / _struct_ref_seq.seq_align_beg / _struct_ref_seq.seq_align_end / _struct_sheet_range.beg_label_seq_id / _struct_sheet_range.end_label_seq_id
解説: Model orientation/position
詳細: correct the orientation on N-terminus of peptide inhibitor
Provider: author / タイプ: Coordinate replacement |
|---|
| 改定 2.1 | 2022年6月29日 | Group: Database references / カテゴリ: citation / citation_author
Item: _citation.country / _citation.journal_abbrev ..._citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.pdbx_database_id_DOI / _citation.title / _citation.year |
|---|
| 改定 2.2 | 2023年11月29日 | Group: Data collection / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond / pdbx_initial_refinement_model |
|---|
| 改定 2.3 | 2024年10月23日 | Group: Structure summary
カテゴリ: pdbx_entry_details / pdbx_modification_feature
Item: _pdbx_entry_details.has_protein_modification |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj
 集合体
集合体


 分子量: 65.409 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Zn / タイプ: SUBJECT OF INVESTIGATION
分子量: 65.409 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Zn / タイプ: SUBJECT OF INVESTIGATION 分子量: 18.015 Da / 分子数: 48 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 48 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: TPS 05A / 波長: 0.99984 Å
/ ビームライン: TPS 05A / 波長: 0.99984 Å 解析
解析