 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7b34 | ||||||
|---|---|---|---|---|---|---|---|
| Title | MST3 in complex with compound MRIA12 | ||||||
 Components Components | Serine/threonine-protein kinase 24 | ||||||
 Keywords Keywords | TRANSFERASE / kinase inhibitors / structure-based drug design / SIK2 inhibitor / Structural Genomics Consortium / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of axon regeneration / Apoptotic execution phase / FAR/SIN/STRIPAK complex / intrinsic apoptotic signaling pathway in response to oxidative stress / execution phase of apoptosis / Apoptotic cleavage of cellular proteins / negative regulation of cell migration / cellular response to starvation / protein autophosphorylation / cellular response to oxidative stress ...regulation of axon regeneration / Apoptotic execution phase / FAR/SIN/STRIPAK complex / intrinsic apoptotic signaling pathway in response to oxidative stress / execution phase of apoptosis / Apoptotic cleavage of cellular proteins / negative regulation of cell migration / cellular response to starvation / protein autophosphorylation / cellular response to oxidative stress / protein phosphorylation / protein kinase activity / non-specific serine/threonine protein kinase / intracellular signal transduction / cadherin binding / protein serine kinase activity / protein serine/threonine kinase activity / nucleolus / Golgi apparatus / signal transduction / extracellular exosome / nucleoplasm / ATP binding / membrane / metal ion binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Tesch, R. / Rak, M. / Joerger, A.C. / Knapp, S. / Structural Genomics Consortium (SGC) | ||||||
| Funding support |  Germany, 1items Germany, 1items
| ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2021 Title: Structure-Based Design of Selective Salt-Inducible Kinase Inhibitors. Authors: Tesch, R. / Rak, M. / Raab, M. / Berger, L.M. / Kronenberger, T. / Joerger, A.C. / Berger, B.T. / Abdi, I. / Hanke, T. / Poso, A. / Strebhardt, K. / Sanhaji, M. / Knapp, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7b34.cif.gz | 144.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7b34.ent.gz | 93.3 KB | Display | PDB format |
| PDBx/mmJSON format | 7b34.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b3/7b34ftp://data.pdbj.org/pub/pdb/validation_reports/b3/7b34 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7b30C  7b31C  7b32C  7b33C  7b35C  7b36C  3zhpS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34559.648 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: STK24, MST3, STK3 / Production host:  References: UniProt: Q9Y6E0, non-specific serine/threonine protein kinase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2#3: Chemical | ChemComp-SQK / | 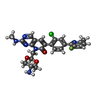  Mass: 510.948 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H24ClFN6O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 510.948 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H24ClFN6O3 / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 66 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 66 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.97 Å3/Da / Density % sol: 58.61 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: Protein solution: 10 mg/ml in buffer 25 mM HEPES pH 7.5, 200 mM NaCl, 5% glycerol, 0.5 mM TCEP. Reservoir: 14% PEG 6000, 0.1M HEPES pH 7.2. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1.00004 Å / Beamline: X06SA / Wavelength: 1.00004 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Mar 7, 2020 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.00004 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→39.73 Å / Num. obs: 20791 / % possible obs: 99.1 % / Redundancy: 5.4 % / Biso Wilson estimate: 37.93 Å2 / CC1/2: 0.997 / Rpim(I) all: 0.042 / Net I/σ(I): 12.1 |
| Reflection shell | Resolution: 2.1→2.16 Å / Redundancy: 5.6 % / Mean I/σ(I) obs: 2.1 / Num. unique obs: 1732 / CC1/2: 0.518 / Rpim(I) all: 0.477 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3ZHP Resolution: 2.1→39.73 Å / SU ML: 0.26 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 24.4887 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 46.97 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→39.73 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
