 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6zpm: Crystal structure of the unconventional kinetochore protein Trypa... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6zpm | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the unconventional kinetochore protein Trypanosoma cruzi KKT4 coiled coil domain | ||||||
 Components Components | Trypanosoma cruzi KKT4 117-218 | ||||||
 Keywords Keywords | CELL CYCLE / KKT4 / kinetochore / kinetoplastids / microtubules | ||||||
| Function / homology | BRCT domain superfamily / THREONINE / Putative kinetoplastid kinetochore protein 4 Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO PHASING / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO PHASING / Resolution: 1.9 Å | ||||||
 Authors Authors | Ludzia, P. / Lowe, D.E. / Marciano, G. / Mohammed, S. / Redfield, C. / Akiyoshi, B. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Structure / Year: 2021 Title: Structural characterization of KKT4, an unconventional microtubule-binding kinetochore protein. Authors: Ludzia, P. / Lowe, E.D. / Marciano, G. / Mohammed, S. / Redfield, C. / Akiyoshi, B. #1: Journal: Biorxiv / Year: 2020Title: Structural characterisation of KKT4, an unconventional microtubule-binding kinetochore protein Authors: Ludzia, P. / Lowe, E. / Marciano, G. / Mohammed, S. / Redfield, C. / Akiyoshi, B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6zpm.cif.gz | 101 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6zpm.ent.gz | 77.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6zpm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zp/6zpmftp://data.pdbj.org/pub/pdb/validation_reports/zp/6zpm | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
|
-Components
| #1: Protein | Mass: 12437.032 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: N-terminal residue Gly is not visible in the electron density. Source: (gene. exp.)  #2: Chemical | ChemComp-THR / | 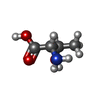  Type: L-peptide linking / Mass: 119.119 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H9NO3 Type: L-peptide linking / Mass: 119.119 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H9NO3#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 184 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 184 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.37 Å3/Da / Density % sol: 48.03 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: HEPES, sodium chloride, MOPSO, Bis-Tris, DL-Arginine hydrochloride, DL- Threonine, DL-Histidine monohydrochloride monohydrate, DL-5-Hydroxylysine hydrochloride, trans-4-hydroxy-L-proline, ...Details: HEPES, sodium chloride, MOPSO, Bis-Tris, DL-Arginine hydrochloride, DL- Threonine, DL-Histidine monohydrochloride monohydrate, DL-5-Hydroxylysine hydrochloride, trans-4-hydroxy-L-proline, PEG 8000, 1,5- Pentanediol, TCEP |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I03 / Wavelength: 0.976 Å |
| Detector | Type: DECTRIS EIGER2 XE 16M / Detector: PIXEL / Date: Feb 22, 2020 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.976 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→67.96 Å / Num. obs: 16787 / % possible obs: 90.36 % / Redundancy: 6.1 % / CC1/2: 0.998 / Rmerge(I) obs: 0.095 / Rpim(I) all: 0.04 / Rrim(I) all: 0.103 / Net I/σ(I): 6.1 |
| Reflection shell | Resolution: 1.9→3.88 Å / Num. unique obs: 1452 / CC1/2: 0.274 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: AB INITIO PHASING / Resolution: 1.9→67.96 Å / SU ML: 0.19 / Cross valid method: THROUGHOUT / σ(F): 1.4 / Phase error: 26.83 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 90.44 Å2 / Biso mean: 37.1107 Å2 / Biso min: 8.96 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.9→67.96 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 12
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
