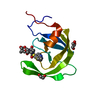
 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6xya | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cap-binding domain of SFTSV L protein | |||||||||
 Components Components | RNA-dependent RNA polymerase | |||||||||
 Keywords Keywords | VIRAL PROTEIN / bunyavirus / cap binding / cap-snatching / viral polymerase | |||||||||
| Function / homology |  Function and homology information Function and homology informationhost cell endoplasmic reticulum / virion component / host cell endoplasmic reticulum-Golgi intermediate compartment / Hydrolases; Acting on ester bonds / host cell Golgi apparatus / RNA-directed RNA polymerase / viral RNA genome replication / hydrolase activity / RNA-directed RNA polymerase activity / DNA-templated transcription / metal ion binding Similarity search - Function | |||||||||
| Biological species |  SFTS virus AH12 SFTS virus AH12 | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.35 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.35 Å | |||||||||
 Authors Authors | Gogrefe, N. / Guenther, S. / Rosenthal, M. | |||||||||
| Funding support |  Germany, European Union, 2items Germany, European Union, 2items
| |||||||||
 Citation Citation | Journal: Nucleic Acids Res / Year: 2020 Title: Structural and functional characterization of the severe fever with thrombocytopenia syndrome virus L protein. Authors: Dominik Vogel / Sigurdur Rafn Thorkelsson / Emmanuelle R J Quemin / Kristina Meier / Tomas Kouba / Nadja Gogrefe / Carola Busch / Sophia Reindl / Stephan Günther / Stephen Cusack / Kay ...Authors: Dominik Vogel / Sigurdur Rafn Thorkelsson / Emmanuelle R J Quemin / Kristina Meier / Tomas Kouba / Nadja Gogrefe / Carola Busch / Sophia Reindl / Stephan Günther / Stephen Cusack / Kay Grünewald / Maria Rosenthal /  Abstract: The Bunyavirales order contains several emerging viruses with high epidemic potential, including Severe fever with thrombocytopenia syndrome virus (SFTSV). The lack of medical countermeasures, such ...The Bunyavirales order contains several emerging viruses with high epidemic potential, including Severe fever with thrombocytopenia syndrome virus (SFTSV). The lack of medical countermeasures, such as vaccines and antivirals, is a limiting factor for the containment of any virus outbreak. To develop such antivirals a profound understanding of the viral replication process is essential. The L protein of bunyaviruses is a multi-functional and multi-domain protein performing both virus transcription and genome replication and, therefore, is an ideal drug target. We established expression and purification procedures for the full-length L protein of SFTSV. By combining single-particle electron cryo-microscopy and X-ray crystallography, we obtained 3D models covering ∼70% of the SFTSV L protein in the apo-conformation including the polymerase core region, the endonuclease and the cap-binding domain. We compared this first L structure of the Phenuiviridae family to the structures of La Crosse peribunyavirus L protein and influenza orthomyxovirus polymerase. Together with a comprehensive biochemical characterization of the distinct functions of SFTSV L protein, this work provides a solid framework for future structural and functional studies of L protein-RNA interactions and the development of antiviral strategies against this group of emerging human pathogens. #1: Journal: Biorxiv / Year: 2020Title: Structural and functional characterization of the Severe fever with thrombocytopenia syndrome virus L protein Authors: Vogel, D. / Thorkelsson, S.R. / Quemin, E. / Meier, K. / Kouba, T. / Gogrefe, N. / Guenther, S. / Cusack, S. / Grunewald, K. / Rosenthal, M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6xya.cif.gz | 108.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6xya.ent.gz | 68.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6xya.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xy/6xyaftp://data.pdbj.org/pub/pdb/validation_reports/xy/6xya | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6y6kC  6qhgS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 12844.638 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: the first two residues (GP) are remains of an enzymatic cleavage siite and not originally part of the sequence Source: (gene. exp.) SFTS virus AH12 / Production host:  |
|---|---|
| #2: Chemical | ChemComp-MGP / 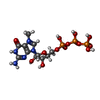  Mass: 538.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H19N5O14P3 / Feature type: SUBJECT OF INVESTIGATION Mass: 538.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H19N5O14P3 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 189 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 189 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.15 Å3/Da / Density % sol: 42.91 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: Final concentrations of components: 16.6 mM Sodium phosphate (pH 6.5), 50mM NaCl, 6.6% (w/v) Glycerol, 4.7% (v/v) 2-Butanol, 100mM Mes (pH 6.0), 18.3% (w/v) Polyethylene glycol 4000, 1.3 mM ...Details: Final concentrations of components: 16.6 mM Sodium phosphate (pH 6.5), 50mM NaCl, 6.6% (w/v) Glycerol, 4.7% (v/v) 2-Butanol, 100mM Mes (pH 6.0), 18.3% (w/v) Polyethylene glycol 4000, 1.3 mM m7GTP Protein concentration: SFTSV CBD 12.4 mg/ml |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY / Beamline: P14 (MX2) / Wavelength: 0.9763 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Oct 22, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 1.35→50 Å / Num. obs: 321482 / % possible obs: 97.59 % / Redundancy: 13.2 % / Biso Wilson estimate: 16.17 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.03094 / Rrim(I) all: 0.03223 / Net I/σ(I): 41.6 |
| Reflection shell | Resolution: 1.35→1.398 Å / Redundancy: 11.6 % / Rmerge(I) obs: 0.3684 / Mean I/σ(I) obs: 6.32 / Num. unique obs: 25283 / CC1/2: 0.953 / Rrim(I) all: 0.3857 / % possible all: 88.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6qhg Resolution: 1.35→33.22 Å / SU ML: 0.1152 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 20.1928 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.67 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.35→33.22 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -12.7327782818 Å / Origin y: -1.59575522964 Å / Origin z: 6.63915482155 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
