protein targeting to vacuole involved in autophagy / vacuole / protein polymerization / autophagosome / ubiquitin binding / zinc ion binding / cytoplasm Similarity search - Function
: / Next to BRCA1 gene 1 protein-like, C-terminal UBA domain / Next to BRCA1, central domain / Ig-like domain from next to BRCA1 gene / PB1 domain / PB1 domain / PB1 domain / Zinc finger, ZZ type / Zinc-binding domain, present in Dystrophin, CREB-binding protein. / Zinc finger, ZZ-type ...: / Next to BRCA1 gene 1 protein-like, C-terminal UBA domain / Next to BRCA1, central domain / Ig-like domain from next to BRCA1 gene / PB1 domain / PB1 domain / PB1 domain / Zinc finger, ZZ type / Zinc-binding domain, present in Dystrophin, CREB-binding protein. / Zinc finger, ZZ-type / Zinc finger, ZZ-type superfamily / Zinc finger ZZ-type profile. / UBA-like superfamily / Immunoglobulin-like fold Similarity search - Domain/homology
Journal: Nat Commun / Year: 2020 Title: Structural basis of p62/SQSTM1 helical filaments and their role in cellular cargo uptake. Authors: Arjen J Jakobi / Stefan T Huber / Simon A Mortensen / Sebastian W Schultz / Anthimi Palara / Tanja Kuhm / Birendra Kumar Shrestha / Trond Lamark / Wim J H Hagen / Matthias Wilmanns / Terje ...Authors: Arjen J Jakobi / Stefan T Huber / Simon A Mortensen / Sebastian W Schultz / Anthimi Palara / Tanja Kuhm / Birendra Kumar Shrestha / Trond Lamark / Wim J H Hagen / Matthias Wilmanns / Terje Johansen / Andreas Brech / Carsten Sachse / Abstract: p62/SQSTM1 is an autophagy receptor and signaling adaptor with an N-terminal PB1 domain that forms the scaffold of phase-separated p62 bodies in the cell. The molecular determinants that govern PB1 ...p62/SQSTM1 is an autophagy receptor and signaling adaptor with an N-terminal PB1 domain that forms the scaffold of phase-separated p62 bodies in the cell. The molecular determinants that govern PB1 domain filament formation in vitro remain to be determined and the role of p62 filaments inside the cell is currently unclear. We here determine four high-resolution cryo-EM structures of different human and Arabidopsis PB1 domain assemblies and observed a filamentous ultrastructure of p62/SQSTM1 bodies using correlative cellular EM. We show that oligomerization or polymerization, driven by a double arginine finger in the PB1 domain, is a general requirement for lysosomal targeting of p62. Furthermore, the filamentous assembly state of p62 is required for autophagosomal processing of the p62-specific cargo KEAP1. Our results show that using such mechanisms, p62 filaments can be critical for cargo uptake in autophagy and are an integral part of phase-separated p62 bodies.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Germany, 3items
Germany, 3items  Citation
Citation

 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

















 PDBj
PDBj





 Assembly
Assembly





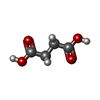

 Mass: 92.094 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4
Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4 Sample preparation
Sample preparation / Beamline: ID23-2 / Wavelength: 0.873 Å
/ Beamline: ID23-2 / Wavelength: 0.873 Å Processing
Processing