Cryo-EM structure of mouse cytoplasmic dynein-1 microtubule binding domain bound to microtubules
Components
MKIAA0325 protein
Tubulin alpha-1B chain
Tubulin beta chain
Keywords
MOTOR PROTEIN / filament / complex
Function / homology
Function and homology information
COPI-independent Golgi-to-ER retrograde traffic / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / Amplification of signal from unattached kinetochores via a MAD2 inhibitory signal / COPI-mediated anterograde transport / cilium movement / Aggrephagy / Mitotic Prometaphase / EML4 and NUDC in mitotic spindle formation / Resolution of Sister Chromatid Cohesion / RHO GTPases Activate Formins ...COPI-independent Golgi-to-ER retrograde traffic / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / Amplification of signal from unattached kinetochores via a MAD2 inhibitory signal / COPI-mediated anterograde transport / cilium movement / Aggrephagy / Mitotic Prometaphase / EML4 and NUDC in mitotic spindle formation / Resolution of Sister Chromatid Cohesion / RHO GTPases Activate Formins / Loss of Nlp from mitotic centrosomes / Recruitment of mitotic centrosome proteins and complexes / Loss of proteins required for interphase microtubule organization from the centrosome / Anchoring of the basal body to the plasma membrane / Separation of Sister Chromatids / Recruitment of NuMA to mitotic centrosomes / AURKA Activation by TPX2 / dynein complex / manchette / Regulation of PLK1 Activity at G2/M Transition / positive regulation of intracellular transport / regulation of metaphase plate congression / positive regulation of spindle assembly / MHC class II antigen presentation / establishment of spindle localization / Microtubule-dependent trafficking of connexons from Golgi to the plasma membrane / Resolution of Sister Chromatid Cohesion / Hedgehog 'off' state / Cilium Assembly / Intraflagellar transport / COPI-dependent Golgi-to-ER retrograde traffic / Mitotic Prometaphase / Carboxyterminal post-translational modifications of tubulin / RHOH GTPase cycle / EML4 and NUDC in mitotic spindle formation / Sealing of the nuclear envelope (NE) by ESCRT-III / Kinesins / PKR-mediated signaling / Separation of Sister Chromatids / The role of GTSE1 in G2/M progression after G2 checkpoint / Aggrephagy / retrograde axonal transport / RHO GTPases activate IQGAPs / RHO GTPases Activate Formins / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / minus-end-directed microtubule motor activity / P-body assembly / MHC class II antigen presentation / Recruitment of NuMA to mitotic centrosomes / dynein light intermediate chain binding / cytoplasmic dynein complex / COPI-mediated anterograde transport / microtubule-based movement / nuclear migration / dynein intermediate chain binding / cytoplasmic microtubule / cytoplasmic microtubule organization / Neutrophil degranulation / axon cytoplasm / stress granule assembly / mitotic spindle organization / regulation of mitotic spindle organization / filopodium / structural constituent of cytoskeleton / microtubule cytoskeleton organization / neuron migration / nuclear envelope / mitotic cell cycle / positive regulation of cold-induced thermogenesis / microtubule cytoskeleton / cell cortex / microtubule / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / axon / cell division / neuronal cell body / GTPase activity / centrosome / GTP binding / ATP binding / metal ion binding / identical protein binding / cytoplasm Similarity search - Function
Dynein heavy chain, AAA 5 extension domain / Dynein heavy chain AAA lid domain / Dynein heavy chain, C-terminal domain / Dynein heavy chain, C-terminal domain, barrel region / Dynein heavy chain C-terminal domain / : / Dynein heavy chain, ATPase lid domain / Dynein heavy chain, tail / Dynein heavy chain, N-terminal region 1 / P-loop containing dynein motor region ...Dynein heavy chain, AAA 5 extension domain / Dynein heavy chain AAA lid domain / Dynein heavy chain, C-terminal domain / Dynein heavy chain, C-terminal domain, barrel region / Dynein heavy chain C-terminal domain / : / Dynein heavy chain, ATPase lid domain / Dynein heavy chain, tail / Dynein heavy chain, N-terminal region 1 / P-loop containing dynein motor region / Dynein heavy chain region D6 P-loop domain / Dynein heavy chain, linker / Dynein heavy chain, AAA module D4 / Dynein heavy chain, coiled coil stalk / Dynein heavy chain / Dynein heavy chain, hydrolytic ATP-binding dynein motor region / Dynein heavy chain, ATP-binding dynein motor region / Dynein heavy chain AAA lid domain / Dynein heavy chain AAA lid domain superfamily / Dynein heavy chain, domain 2, N-terminal / Dynein heavy chain, linker, subdomain 3 / Dynein heavy chain, AAA1 domain, small subdomain / Dynein heavy chain region D6 P-loop domain / Dynein heavy chain, N-terminal region 2 / Hydrolytic ATP binding site of dynein motor region / Microtubule-binding stalk of dynein motor / P-loop containing dynein motor region D4 / ATP-binding dynein motor region / Dynein heavy chain AAA lid domain / Alpha tubulin / Tubulin-beta mRNA autoregulation signal. / Beta tubulin, autoregulation binding site / Beta tubulin / Tubulin / Tubulin, C-terminal / Tubulin C-terminal domain / Tubulin, conserved site / Tubulin subunits alpha, beta, and gamma signature. / Tubulin/FtsZ family, C-terminal domain / Tubulin/FtsZ-like, C-terminal domain / Tubulin/FtsZ, C-terminal / Tubulin/FtsZ, 2-layer sandwich domain / Tubulin/FtsZ family, GTPase domain / Tubulin/FtsZ family, GTPase domain / Tubulin/FtsZ, GTPase domain / Tubulin/FtsZ, GTPase domain superfamily / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / P-loop containing nucleoside triphosphate hydrolase Similarity search - Domain/homology
Journal: Elife / Year: 2019 Title: Cryo-EM of dynein microtubule-binding domains shows how an axonemal dynein distorts the microtubule. Authors: Samuel E Lacey / Shaoda He / Sjors Hw Scheres / Andrew P Carter / Abstract: Dyneins are motor proteins responsible for transport in the cytoplasm and the beating of axonemes in cilia and flagella. They bind and release microtubules via a compact microtubule-binding domain ...Dyneins are motor proteins responsible for transport in the cytoplasm and the beating of axonemes in cilia and flagella. They bind and release microtubules via a compact microtubule-binding domain (MTBD) at the end of a coiled-coil stalk. We address how cytoplasmic and axonemal dynein MTBDs bind microtubules at near atomic resolution. We decorated microtubules with MTBDs of cytoplasmic dynein-1 and axonemal dynein DNAH7 and determined their cryo-EM structures using helical Relion. The majority of the MTBD is rigid upon binding, with the transition to the high-affinity state controlled by the movement of a single helix at the MTBD interface. DNAH7 contains an 18-residue insertion, found in many axonemal dyneins, that contacts the adjacent protofilament. Unexpectedly, we observe that DNAH7, but not dynein-1, induces large distortions in the microtubule cross-sectional curvature. This raises the possibility that dynein coordination in axonemes is mediated via conformational changes in the microtubule.
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors United Kingdom, 2items
United Kingdom, 2items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj











 Assembly
Assembly




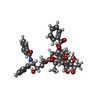
 Mass: 523.180 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM
Mass: 523.180 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM
Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM Mass: 853.906 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C47H51NO14 / Comment: medication, chemotherapy*YM
Mass: 853.906 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C47H51NO14 / Comment: medication, chemotherapy*YM Sample preparation
Sample preparation Electron microscopy imaging
Electron microscopy imaging
 FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM
FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM Processing
Processing