Entry Database : PDB / ID : 6n8yTitle Hsp90-beta bound to PU-11-trans Heat shock protein HSP 90-beta Keywords / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 1.55390799533 Å Authors Huck, J.D. / Que, N.L.S. / Gewirth, D.T. Funding support Organization Grant number Country National Institutes of Health/National Cancer Institute (NIH/NCI) P01CA186866 National Institutes of Health/National Cancer Institute (NIH/NCI) R01CA095130
Journal : Proteins / Year : 2019Title : Structures of Hsp90 alpha and Hsp90 beta bound to a purine-scaffold inhibitor reveal an exploitable residue for drug selectivity.Authors : Huck, J.D. / Que, N.L.S. / Sharma, S. / Taldone, T. / Chiosis, G. / Gewirth, D.T. History Deposition Nov 30, 2018 Deposition site / Processing site Revision 1.0 Jul 3, 2019 Provider / Type Revision 1.1 Jul 10, 2019 Group / Database references / Category / citation_authorItem / _citation.pdbx_database_id_PubMed / _citation.titleRevision 1.2 Sep 11, 2019 Group / Database references / Category Item / _citation.page_first / _citation.page_lastRevision 1.3 Dec 4, 2019 Group / Category / Item Revision 1.4 Oct 11, 2023 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 2items
United States, 2items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads























 PDBj
PDBj
















 Assembly
Assembly

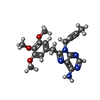
 Mass: 369.418 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H23N5O3 / Feature type: SUBJECT OF INVESTIGATION
Mass: 369.418 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H23N5O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 345 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 345 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing