 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6l7h | ||||||
|---|---|---|---|---|---|---|---|
| Title | crystal structure of GgCGT in complex with UDP and Nothofagin | ||||||
 Components Components | GgCGT1 | ||||||
 Keywords Keywords | TRANSFERASE / di-C-glycosyltransferase | ||||||
| Function / homology | Glycogen Phosphorylase B; / Rossmann fold / 3-Layer(aba) Sandwich / Alpha Beta / Chem-E7F / URIDINE-5'-DIPHOSPHATE Function and homology information Function and homology information | ||||||
| Biological species |  Glycyrrhiza glabra (plant) Glycyrrhiza glabra (plant) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Zhang, M. / Li, F.D. / Ye, M. | ||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2020 Title: Functional Characterization and Structural Basis of an Efficient Di-C-glycosyltransferase fromGlycyrrhiza glabra. Authors: Zhang, M. / Li, F.D. / Li, K. / Wang, Z.L. / Wang, Y.X. / He, J.B. / Su, H.F. / Zhang, Z.Y. / Chi, C.B. / Shi, X.M. / Yun, C.H. / Zhang, Z.Y. / Liu, Z.M. / Zhang, L.R. / Yang, D.H. / Ma, M. / Qiao, X. / Ye, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6l7h.cif.gz | 119.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6l7h.ent.gz | 88.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6l7h.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l7/6l7hftp://data.pdbj.org/pub/pdb/validation_reports/l7/6l7h | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6l5pSC  6l5qC  6l5rC  6l5sC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 52093.066 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Glycyrrhiza glabra (plant) / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | 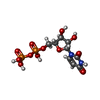  Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM#3: Chemical | ChemComp-E7F / | 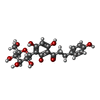  Mass: 436.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H24O10 Mass: 436.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H24O10#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 559 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 559 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.04 Å3/Da / Density % sol: 39.72 % |
|---|---|
| Crystal grow | Temperature: 283 K / Method: vapor diffusion, hanging drop Details: 0.2 M Ammonium acetate, 0.1 M BIS-Tris, pH 6.5, 25% w/v Polyethylene glycol 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 / Wavelength: 1.54056 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 200K / Detector: PIXEL / Date: Oct 31, 2019 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54056 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.8→59.838 Å / Num. all: 39867 / Num. obs: 39867 / % possible obs: 99 % / Redundancy: 6.4 % / Rpim(I) all: 0.041 / Rrim(I) all: 0.107 / Rsym value: 0.098 / Net I/av σ(I): 6.8 / Net I/σ(I): 12.5 / Num. measured all: 256181 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6L5P Resolution: 1.8→9.002 Å / SU ML: 0.18 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 21.06
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 91.56 Å2 / Biso mean: 19.5475 Å2 / Biso min: 3.53 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.8→9.002 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 14
|
