| Entry | Database: PDB / ID: 6if4
|
|---|
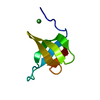
| Title | Crystal structure of Tbtudor |
|---|
 Components Components | Histone acetyltransferase |
|---|
 Keywords Keywords | DNA BINDING PROTEIN / Tudor / DNA / transcription / methylation |
|---|
| Function / homology |  Function and homology information Function and homology information
histone acetyltransferase activity / histone acetyltransferase / transcription coregulator activity / DNA repair / chromatin binding / regulation of transcription by RNA polymerase II / chromatin / DNA binding / nucleusSimilarity search - Function : / Histone acetyltransferase domain, MYST-type / MOZ/SAS family / MYST-type histone acetyltransferase (HAT) domain profile. / RNA binding activity-knot of a chromodomain / RNA binding activity-knot of a chromodomain / Chromo-like domain superfamily / Acyl-CoA N-acyltransferase / Winged helix-like DNA-binding domain superfamilySimilarity search - Domain/homology |
|---|
| Biological species |   Trypanosoma brucei brucei (eukaryote) Trypanosoma brucei brucei (eukaryote) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.934 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.934 Å |
|---|
 Authors Authors | Gao, J. / Ye, K. / Diwu, Y. / Liao, S. / Tu, X. |
|---|
| Funding support |  China, 3items China, 3items | Organization | Grant number | Country |
|---|
| National Natural Science Foundation of China | 31500601 | China | | National Natural Science Foundation of China | U1332137 | China | | National Natural Science Foundation of China | 31570737 | China |
|
|---|
 Citation Citation | Journal: J.Struct.Biol. / Year: 2020
Title: Crystal structure of TbEsa1 presumed Tudor domain from Trypanosoma brucei.
Authors: Gao, J. / Ye, K. / Diwu, Y. / Xu, C. / Zhang, X. / Liao, S. / Tu, X. |
|---|
| History | | Deposition | Sep 18, 2018 | Deposition site: PDBJ / Processing site: PDBJ |
|---|
| Revision 1.0 | Sep 18, 2019 | Provider: repository / Type: Initial release |
|---|
| Revision 2.0 | Nov 6, 2019 | Group: Advisory / Atomic model ...Advisory / Atomic model / Data collection / Derived calculations / Other / Refinement description / Structure summary
Category: atom_site / atom_site_anisotrop ...atom_site / atom_site_anisotrop / atom_sites / computing / diffrn / entity / pdbx_nonpoly_scheme / pdbx_refine_tls / pdbx_refine_tls_group / pdbx_struct_assembly_gen / pdbx_unobs_or_zero_occ_atoms / pdbx_validate_close_contact / pdbx_validate_rmsd_angle / pdbx_validate_rmsd_bond / pdbx_validate_symm_contact / pdbx_validate_torsion / refine / refine_hist / refine_ls_restr / refine_ls_restr_ncs / refine_ls_shell / software / struct_ncs_dom / struct_ncs_dom_lim / struct_sheet_range
Item: _atom_sites.fract_transf_matrix[1][2] / _atom_sites.fract_transf_matrix[1][3] ..._atom_sites.fract_transf_matrix[1][2] / _atom_sites.fract_transf_matrix[1][3] / _atom_sites.fract_transf_matrix[2][3] / _atom_sites.fract_transf_matrix[3][3] / _diffrn.pdbx_serial_crystal_experiment / _entity.pdbx_number_of_molecules / _pdbx_struct_assembly_gen.asym_id_list / _refine.B_iso_max / _refine.B_iso_mean / _refine.B_iso_min / _refine.ls_R_factor_R_free / _refine.ls_R_factor_R_work / _refine.ls_R_factor_obs / _refine.ls_number_reflns_R_work / _refine.overall_SU_ML / _refine.pdbx_ls_cross_valid_method / _refine.pdbx_overall_phase_error / _refine.pdbx_stereochemistry_target_values / _refine.solvent_model_details / _refine_hist.cycle_id / _refine_hist.number_atoms_solvent / _refine_hist.number_atoms_total / _refine_hist.pdbx_B_iso_mean_solvent / _refine_hist.pdbx_number_atoms_protein / _refine_hist.pdbx_number_residues_total / _refine_ls_shell.R_factor_R_free / _refine_ls_shell.R_factor_R_free_error / _refine_ls_shell.R_factor_R_work / _refine_ls_shell.d_res_high / _refine_ls_shell.d_res_low / _software.version / _struct_sheet_range.end_auth_comp_id / _struct_sheet_range.end_auth_seq_id / _struct_sheet_range.end_label_comp_id / _struct_sheet_range.end_label_seq_id
Description: Atomic clashes / Provider: author / Type: Coordinate replacement |
|---|
| Revision 2.1 | Sep 30, 2020 | Group: Database references / Category: citation / citation_author
Item: _citation.country / _citation.journal_abbrev ..._citation.country / _citation.journal_abbrev / _citation.journal_id_ASTM / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year |
|---|
| Revision 2.2 | Nov 22, 2023 | Group: Data collection / Database references / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / struct_ncs_dom_lim
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ncs_dom_lim.beg_auth_comp_id / _struct_ncs_dom_lim.beg_label_asym_id / _struct_ncs_dom_lim.beg_label_comp_id / _struct_ncs_dom_lim.beg_label_seq_id / _struct_ncs_dom_lim.end_auth_comp_id / _struct_ncs_dom_lim.end_label_asym_id / _struct_ncs_dom_lim.end_label_comp_id / _struct_ncs_dom_lim.end_label_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj





 Assembly
Assembly

