 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ia9 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | urate oxidase under 2000 bar (220 MPa) of argon | ||||||||||||
 Components Components | Uricase | ||||||||||||
 Keywords Keywords | OXIDOREDUCTASE / aspergillus flavus / homotetramer / purine metabolism / argon / high pressure | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationfactor-independent urate hydroxylase / urate oxidase activity / purine nucleobase catabolic process / urate catabolic process / peroxisome Similarity search - Function | ||||||||||||
| Biological species |  | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||||||||
 Authors Authors | Prange, T. / Colloc'h, N. / Carpentier, P. | ||||||||||||
| Funding support | 1items
| ||||||||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2022 Title: Comparative study of the effects of high hydrostatic pressure per se and high argon pressure on urate oxidase ligand stabilization. Authors: Prange, T. / Carpentier, P. / Dhaussy, A.C. / van der Linden, P. / Girard, E. / Colloc'h, N. #1: Journal: Current trends in X-ray crystallography / Year: 2011Title: Current trends in X-ray Crystallography. Intech Open books Authors: Colloc'h, N. / Marassio, G. / Prange, T. #2: Journal: Journal of Applied Crystallography / Year: 2016Title: Gas-sensitive biological crystals processed in pressurized oxygen and krypton atmospheres: deciphering gas channels in proteins using a novel soak-and-freeze methodology Authors: Lafumat, B. / Mueller-Dieckmann, C. / Leonard, G. / Colloc'h, N. / Prange, T. / Giraud, T. / Dobias, F. / Royant, A. / van der Linden, P. / Carpentier, P. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ia9.cif.gz | 152.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ia9.ent.gz | Display | PDB format | |
| PDBx/mmJSON format | 6ia9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ia/6ia9ftp://data.pdbj.org/pub/pdb/validation_reports/ia/6ia9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6i9xC  6i9zC  6ia1C  6ia3C  7p0cC  7p0dC  7p0gC  7pufC  7pwnC  7q09C  1r56S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules aaaaba
| #1: Protein | Mass: 33498.719 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Komagataella pastoris (fungus)References: UniProt: Q00511, factor-independent urate hydroxylase |
|---|
-Non-polymers , 5 types, 503 molecules 

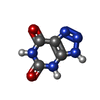


| #2: Chemical |  Mass: 44.053 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H4O Mass: 44.053 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H4O#3: Chemical |  Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#4: Chemical | ChemComp-AZA /  Mass: 153.099 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H3N5O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 153.099 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H3N5O2 / Feature type: SUBJECT OF INVESTIGATION#5: Chemical | ChemComp-AR /  Mass: 39.948 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ar Mass: 39.948 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ar#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 491 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 57.5 % / Description: LARGE COLORLESS PRISMS |
|---|---|
| Crystal grow | Temperature: 291 K / Method: batch mode / pH: 8 Details: 20 microliter of protein (15 mg/ml) mixed with 20 microliter of solution: Buffer Tris 0.05M (chloride free) + 4% PEG 4000. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID30B / Wavelength: 1.77 Å / Beamline: ID30B / Wavelength: 1.77 Å |
| Detector | Type: DECTRIS PILATUS3 S 6M / Detector: PIXEL / Date: May 10, 2016 |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.77 Å / Relative weight: 1 |
| Reflection | Resolution: 1.723→71.322 Å / Num. obs: 72278 / % possible obs: 93.8 % / Redundancy: 11.4 % / CC1/2: 0.999 / Rmerge(I) obs: 0.057 / Rpim(I) all: 0.017 / Rrim(I) all: 0.062 / Net I/σ(I): 22.8 |
| Reflection shell | Resolution: 1.723→1.826 Å / Redundancy: 5.3 % / Rmerge(I) obs: 0.542 / Mean I/σ(I) obs: 2 / Num. unique obs: 3531 / CC1/2: 0.802 / Rpim(I) all: 0.26 / Rrim(I) all: 0.63 / % possible all: 56.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1r56 reconstructed dimer Resolution: 1.8→46.333 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.947 / SU B: 4.014 / SU ML: 0.115 / Cross valid method: FREE R-VALUE / ESU R: 0.13 / ESU R Free: 0.126 / Details: Hydrogens have not been used
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.255 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→46.333 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
|
