 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1r4u: URATE OXIDASE FROM ASPERGILLUS FLAVUS COMPLEXED WITH ITS INHIBITO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1r4u | ||||||
|---|---|---|---|---|---|---|---|
| Title | URATE OXIDASE FROM ASPERGILLUS FLAVUS COMPLEXED WITH ITS INHIBITOR OXONIC ACID | ||||||
 Components Components | Uricase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / URIC ACID DEGRADATION / DIMERIC BARREL / TUNNEL-SHAPED PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationfactor-independent urate hydroxylase / urate oxidase activity / purine nucleobase catabolic process / urate catabolic process / peroxisome Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å | ||||||
 Authors Authors | Retailleau, P. / Colloc'h, N. / Prange, T. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2004 Title: Complexed and ligand-free high-resolution structures of urate oxidase (Uox) from Aspergillus flavus: a reassignment of the active-site binding mode. Authors: Retailleau, P. / Colloc'h, N. / Vivares, D. / Bonnete, F. / Castro, B. / El-Hajji, M. / Mornon, J.P. / Monard, G. / Prange, T. #1: Journal: Nat.Struct.Biol. / Year: 1997Title: Crystal Structure of the Protein Drug Urate Oxidase-Inhibitor Complex at 2.05 A Resolution Authors: Colloc'h, N. / El Hajji, M. / Bachet, B. / L'Hermite, G. / Schiltz, M. / Prange, T. / Castro, B. / Mornon, J.-P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1r4u.cif.gz | 77.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1r4u.ent.gz | 57 KB | Display | PDB format |
| PDBx/mmJSON format | 1r4u.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/r4/1r4uftp://data.pdbj.org/pub/pdb/validation_reports/r4/1r4u | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1r4sC  1r51C  1r56C  1uox C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34199.586 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) References: UniProt: Q00511, factor-independent urate hydroxylase |
|---|---|
| #2: Chemical | ChemComp-OXC / 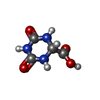  Mass: 159.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H5N3O4 Mass: 159.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H5N3O4 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.78 Å3/Da / Density % sol: 55.3 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 8 Details: 8.5MG/ML PROTEIN, 0.2MG/ML OXONIC ACID, 5-7% W/V PEG 8000, 100MM TRIS/HCL, pH 8.0, VAPOR DIFFUSION, SITTING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 283 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: LURE  / Beamline: DW32 / Wavelength: 0.97 / Wavelength: 0.972 Å / Beamline: DW32 / Wavelength: 0.97 / Wavelength: 0.972 Å | |||||||||
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE | |||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 1.65→20 Å / Num. obs: 48706 / % possible obs: 99.1 % / Redundancy: 8.2 % / Rmerge(I) obs: 0.064 / Net I/σ(I): 13 | |||||||||
| Reflection shell | Resolution: 1.65→1.69 Å / Rmerge(I) obs: 0.375 / Mean I/σ(I) obs: 2.9 / % possible all: 95.9 |
- Processing
Processing
| Software |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1UOX 1uox Resolution: 1.65→20 Å
| |||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.65→20 Å
| |||||||||||||||
| Refine LS restraints |
|
