 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1xt4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Urate Oxidase From Aspergillus Flavus Complexed With Guanine | ||||||
 Components Components | Uricase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / URIC ACID DEGRADATION / DIMERIC BARREL / TUNNEL-SHAPED PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationfactor-independent urate hydroxylase / urate oxidase activity / purine nucleobase catabolic process / urate catabolic process / peroxisome Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.01 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.01 Å | ||||||
 Authors Authors | Retailleau, P. / Colloc'h, N. / Vivares, D. / Bonnete, F. / Castro, B. / El Hajji, M. / Prange, T. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2005 Title: Urate oxidase from Aspergillus flavus: new crystal-packing contacts in relation to the content of the active site. Authors: Retailleau, P. / Colloc'h, N. / Vivares, D. / Bonnete, F. / Castro, B. / El Hajji, M. / Prange, T. #1: Journal: Nat.Struct.Biol. / Year: 1997 Title: Crystal structure of the protein drug urate oxidase-inhibitor complex at 2.05 A resolution Authors: Colloc'h, N. / El Hajji, M. / Bachet, B. / L'Hermite, G. / Schiltz, M. / Castro, B. / Mornon, J.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1xt4.cif.gz | 75.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1xt4.ent.gz | 55.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1xt4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xt/1xt4ftp://data.pdbj.org/pub/pdb/validation_reports/xt/1xt4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1wrrC  1ws2C  1ws3C  1xxjC  1xy3C  1r51S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a tetramer generated from the monomer in the asymmetric unit by the operations: x, 1-y, 1-z, and 1-x, y, 1-z and 1-x, 1-y, z |
-Components
| #1: Protein | Mass: 34199.586 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: Q00511, factor-independent urate hydroxylase |
|---|---|
| #2: Chemical | ChemComp-GUN / 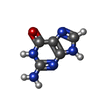  Mass: 151.126 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H5N5O Mass: 151.126 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H5N5O |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 117 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 117 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.88 Å3/Da / Density % sol: 56.9 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 8 Details: 8.5MG/ML PROTEIN, 0.2MG/ML Guanine, 5-7%(W/V) PEG 8000, 100MM TRIS/HCL, pH 8.0, VAPOR DIFFUSION, SITTING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Nov 23, 2003 / Details: mirrors |
| Radiation | Monochromator: OSMIC MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→25 Å / Num. obs: 26240 / % possible obs: 94.1 % / Redundancy: 5.5 % / Biso Wilson estimate: 33.1 Å2 / Rsym value: 0.134 / Net I/σ(I): 13.5 |
| Reflection shell | Resolution: 2→2.15 Å / Mean I/σ(I) obs: 9.4 / Rsym value: 0.281 / % possible all: 90.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1r51 Resolution: 2.01→15 Å / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 29.3 Å2 | ||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.205 Å | ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.01→15 Å
| ||||||||||||||||||||
| Refine LS restraints |
|
