 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7a0l: Joint neutron/X-ray room temperature structure of perdeuterated A... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7a0l | ||||||
|---|---|---|---|---|---|---|---|
| Title | Joint neutron/X-ray room temperature structure of perdeuterated Aspergillus flavus urate oxidase in complex with the 8-azaxanthine inhibitor and catalytic water bound in the peroxo hole | ||||||
 Components Components | Uricase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / COFACTOR-FREE OXIDASE / INHIBITOR / CATALYTIC WATER / DIOXYGEN BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationfactor-independent urate hydroxylase / urate oxidase activity / purine nucleobase catabolic process / urate catabolic process / peroxisome Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / NEUTRON DIFFRACTION / NUCLEAR REACTOR / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.33 Å X-RAY DIFFRACTION / NEUTRON DIFFRACTION / NUCLEAR REACTOR / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.33 Å | ||||||
 Authors Authors | McGregor, L. / Bui, S. / Blakeley, M.P. / Steiner, R.A. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Iucrj / Year: 2021 Title: Joint neutron/X-ray crystal structure of a mechanistically relevant complex of perdeuterated urate oxidase and simulations provide insight into the hydration step of catalysis. Authors: McGregor, L. / Foldes, T. / Bui, S. / Moulin, M. / Coquelle, N. / Blakeley, M.P. / Rosta, E. / Steiner, R.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7a0l.cif.gz | 257.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7a0l.ent.gz | 187.1 KB | Display | PDB format |
| PDBx/mmJSON format | 7a0l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a0/7a0lftp://data.pdbj.org/pub/pdb/validation_reports/a0/7a0l | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4d12S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 34288.742 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: Q00511, factor-independent urate hydroxylase |
|---|---|
| #2: Chemical | ChemComp-AZA / 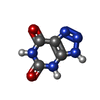  Mass: 153.099 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H3N5O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 153.099 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H3N5O2 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 351 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 351 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment |
|
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.98 Å3/Da / Density % sol: 58.7 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: batch mode / pH: 7.6 / Details: TRIS-Acetate, PEG 8000, 8AZA |
-Data collection
| Diffraction |
| |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| |||||||||||||||||||||||||||
| Detector |
| |||||||||||||||||||||||||||
| Radiation |
| |||||||||||||||||||||||||||
| Radiation wavelength |
| |||||||||||||||||||||||||||
| Reflection | Biso Wilson estimate: 16.6 Å2 / Entry-ID: 7A0L
| |||||||||||||||||||||||||||
| Reflection shell |
|

- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.33→35.51 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
