Entry Database : PDB / ID : 6hpuTitle Crystal structure of human Pif1 helicase in complex with ADP-AlF4 ATP-dependent DNA helicase PIF1 Keywords / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 3.96 Å Authors Levdikov, V.M. / Dehghani-Tafti, S. / Bax, B.D. / Sanders, C.M. / Antson, A.A. Journal : Nucleic Acids Res. / Year : 2019Title : Structural and functional analysis of the nucleotide and DNA binding activities of the human PIF1 helicase.Authors : Dehghani-Tafti, S. / Levdikov, V. / Antson, A.A. / Bax, B. / Sanders, C.M. History Deposition Sep 21, 2018 Deposition site / Processing site Revision 1.0 Jan 23, 2019 Provider / Type Revision 1.1 Feb 13, 2019 Group / Database references / Category / citation_author / pdbx_database_procItem _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_ASTM / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.year / _citation_author.identifier_ORCID / _citation_author.name Revision 1.2 Apr 17, 2019 Group / Database references / Category / pdbx_database_procItem / _citation.page_first / _citation.page_lastRevision 1.3 Jan 24, 2024 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj




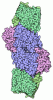


 Assembly
Assembly




 Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM
Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM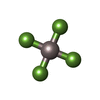
 Mass: 102.975 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: AlF4
Mass: 102.975 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: AlF4
 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Sample preparation
Sample preparation / Beamline: I04 / Wavelength: 0.92819 Å
/ Beamline: I04 / Wavelength: 0.92819 Å Processing
Processing