positive regulation of endodeoxyribonuclease activity / regulation of tRNA methylation / negative regulation of protein maturation / negative regulation of fatty acid beta-oxidation / positive regulation of protein localization to endoplasmic reticulum / regulation of glycogen biosynthetic process / negative regulation of lymphocyte migration / cellular response to decreased oxygen levels / cellular response to rapamycin / negative regulation of protein localization to lysosome ...positive regulation of endodeoxyribonuclease activity / regulation of tRNA methylation / negative regulation of protein maturation / negative regulation of fatty acid beta-oxidation / positive regulation of protein localization to endoplasmic reticulum / regulation of glycogen biosynthetic process / negative regulation of lymphocyte migration / cellular response to decreased oxygen levels / cellular response to rapamycin / negative regulation of protein localization to lysosome / maternal placenta development / maintenance of protein location in mitochondrion / mammalian oogenesis stage / AKT-mediated inactivation of FOXO1A / negative regulation of long-chain fatty acid import across plasma membrane / Negative regulation of the PI3K/AKT network / regulation of type B pancreatic cell development / positive regulation of anaphase-promoting complex-dependent catabolic process / : / potassium channel activator activity / AKT phosphorylates targets in the nucleus / activation-induced cell death of T cells / cellular response to oxidised low-density lipoprotein particle stimulus / negative regulation of cilium assembly / negative regulation of hydrogen peroxide-induced neuron intrinsic apoptotic signaling pathway / Butyrate Response Factor 1 (BRF1) binds and destabilizes mRNA / positive regulation of TORC2 signaling / positive regulation of glucose metabolic process / RUNX2 regulates genes involved in cell migration / cellular response to peptide / beta-arrestin-dependent dopamine receptor signaling pathway / positive regulation of organ growth / mammary gland epithelial cell differentiation / fibroblast migration / interleukin-18-mediated signaling pathway / positive regulation of sodium ion transport / MTOR signalling / response to growth factor / response to fluid shear stress / cellular response to granulocyte macrophage colony-stimulating factor stimulus / negative regulation of leukocyte cell-cell adhesion / RAB GEFs exchange GTP for GDP on RABs / glycogen biosynthetic process / peripheral nervous system myelin maintenance / cell migration involved in sprouting angiogenesis / phosphatidylinositol-3,4-bisphosphate binding / positive regulation of protein localization to cell surface / phosphorylation / complement receptor mediated signaling pathway / sphingosine-1-phosphate receptor signaling pathway / AKT phosphorylates targets in the cytosol / anoikis / regulation of postsynapse organization / response to growth hormone / positive regulation of fibroblast migration / labyrinthine layer blood vessel development / execution phase of apoptosis / regulation of myelination / response to UV-A / KSRP (KHSRP) binds and destabilizes mRNA / Regulation of TP53 Activity through Association with Co-factors / sperm glycocalyx / peptidyl-threonine phosphorylation / TORC2 complex binding / Mechanical load activates signaling by PIEZO1 and integrins in osteocytes / Co-inhibition by CTLA4 / perinuclear theca / negative regulation of cGAS/STING signaling pathway / negative regulation of macroautophagy / cellular response to stress / negative regulation of release of cytochrome c from mitochondria / regulation of neuron projection development / response to food / negative regulation of PERK-mediated unfolded protein response / Constitutive Signaling by AKT1 E17K in Cancer / apoptotic mitochondrial changes / phosphatidylinositol-3,4,5-trisphosphate binding / positive regulation of protein metabolic process / Regulation of localization of FOXO transcription factors / cellular response to vascular endothelial growth factor stimulus / behavioral response to pain / TOR signaling / negative regulation of Notch signaling pathway / positive regulation of peptidyl-serine phosphorylation / CD28 dependent PI3K/Akt signaling / Activation of BAD and translocation to mitochondria / positive regulation of blood vessel endothelial cell migration / Estrogen-dependent nuclear events downstream of ESR-membrane signaling / positive regulation of glycogen biosynthetic process / Mitochondrial unfolded protein response (UPRmt) / negative regulation of extrinsic apoptotic signaling pathway in absence of ligand / protein serine/threonine kinase inhibitor activity / response to insulin-like growth factor stimulus / SARS-CoV-2 targets host intracellular signalling and regulatory pathways / positive regulation of lipid biosynthetic process / positive regulation of fat cell differentiation / negative regulation of oxidative stress-induced intrinsic apoptotic signaling pathway / Cyclin E associated events during G1/S transition / positive regulation of G1/S transition of mitotic cell cycle / vascular endothelial cell response to laminar fluid shear stress Similarity search - Function
Protein kinase B alpha, catalytic domain / Protein Kinase B, pleckstrin homology domain / Protein kinase, C-terminal / Protein kinase C terminal domain / Pleckstrin-homology domain (PH domain)/Phosphotyrosine-binding domain (PTB) / PH-domain like / Extension to Ser/Thr-type protein kinases / AGC-kinase, C-terminal / AGC-kinase C-terminal domain profile. / PH domain ...Protein kinase B alpha, catalytic domain / Protein Kinase B, pleckstrin homology domain / Protein kinase, C-terminal / Protein kinase C terminal domain / Pleckstrin-homology domain (PH domain)/Phosphotyrosine-binding domain (PTB) / PH-domain like / Extension to Ser/Thr-type protein kinases / AGC-kinase, C-terminal / AGC-kinase C-terminal domain profile. / PH domain / PH domain profile. / Pleckstrin homology domain. / Pleckstrin homology domain / PH-like domain superfamily / Roll / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Beta / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.18 Å3/Da / Density % sol: 43.63 %
Crystal grow
Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.2 Details: 1.25 mM Na-acetate, 3.75 mM Na-citrate, 15% v/v PEG 2000 MME, pH 5.2, 3 mg/mL Akt1 (in 25 mM TRIS, 100 mM NaCl, 10 % Glycerol, 5 mM DTT, pH 7.5), 1 uL reservoir + 1 uL protein solution
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Germany, 2items
Germany, 2items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj




























 Assembly
Assembly

 Spodoptera frugiperda (fall armyworm)
Spodoptera frugiperda (fall armyworm)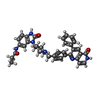
 Mass: 598.694 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C36H34N6O3
Mass: 598.694 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C36H34N6O3 Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X10SA / Wavelength: 0.97862 Å
/ Beamline: X10SA / Wavelength: 0.97862 Å Processing
Processing