 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6he3: Pseudomonas aeruginosa Seryl-tRNA Synthetase in Complex with 5'-O... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6he3 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Pseudomonas aeruginosa Seryl-tRNA Synthetase in Complex with 5'-O-(N-(L-seryl)-sulfamoyl)cytidine | ||||||||||||
 Components Components | Serine--tRNA ligase | ||||||||||||
 Keywords Keywords | LIGASE / Coil-coil / Beta barrel / tRNA Synthetase / Inhibitor / Complex | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationL-selenocysteine biosynthetic process / serine-tRNA ligase / serine-tRNA ligase activity / seryl-tRNA aminoacylation / ATP binding / cytoplasm / cytosol Similarity search - Function | ||||||||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.16 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.16 Å | ||||||||||||
 Authors Authors | Pang, L. / De Graef, S. / Strelkov, S.V. / Weeks, S.D. | ||||||||||||
| Funding support |  Belgium, 3items Belgium, 3items
| ||||||||||||
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2020 Title: Structural Insights into the Binding of Natural Pyrimidine-Based Inhibitors of Class II Aminoacyl-tRNA Synthetases. Authors: Pang, L. / Nautiyal, M. / De Graef, S. / Gadakh, B. / Zorzini, V. / Economou, A. / Strelkov, S.V. / Van Aerschot, A. / Weeks, S.D. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6he3.cif.gz | 327.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6he3.ent.gz | 263.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6he3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/he/6he3ftp://data.pdbj.org/pub/pdb/validation_reports/he/6he3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6hdzC  6he1C  6hhvC  6hhwC  6hhxC  6hhyC  6hhzC  6hi0C  6rltC  6rluC  6rlvC  6s30C  6sjcC  2dq3S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 47305.516 Da / Num. of mol.: 2 / Fragment: Seryl-tRNA Synthetase Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria) / Gene: serS, ALP65_01227 / Plasmid: pETRUK / Production host: References: UniProt: A0A3P3Q1W7, UniProt: Q9I0M6*PLUS, serine-tRNA ligase #2: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H6O2#3: Chemical | 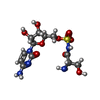  Mass: 409.372 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N5O9S / Feature type: SUBJECT OF INVESTIGATION Mass: 409.372 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N5O9S / Feature type: SUBJECT OF INVESTIGATION#4: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 343 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 343 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 57.61 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8 Details: Holo protein, concentrated to 10 mg/mL in 10 mM Tris pH 7, 100 mM NaCl, 5mM DTT, was mixed with an equal volume of 100 mM Tris pH 8, 200 mM NaCl, 23% w/v PEG 3350 and 5% v/v ethylene glycol. ...Details: Holo protein, concentrated to 10 mg/mL in 10 mM Tris pH 7, 100 mM NaCl, 5mM DTT, was mixed with an equal volume of 100 mM Tris pH 8, 200 mM NaCl, 23% w/v PEG 3350 and 5% v/v ethylene glycol. Suitable crystals were soaked with 2 mM 5'-O-(N-(L-seryl)-sulfamoyl)N3-cytidine in a solution similar to the crystallization condition but containing 22% v/v ethylene glycol. |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-1 / Wavelength: 0.972422 Å / Beamline: ID23-1 / Wavelength: 0.972422 Å | ||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Dec 17, 2017 | ||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.972422 Å / Relative weight: 1 | ||||||||||||||||||||||||
| Reflection | Resolution: 2.1→91.25 Å / Num. obs: 62738 / % possible obs: 99.3 % / Redundancy: 3.4 % / Biso Wilson estimate: 51 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.056 / Rpim(I) all: 0.036 / Rrim(I) all: 0.067 / Net I/σ(I): 11.7 / Num. measured all: 214186 / Scaling rejects: 8 | ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Model generated using 2DQ3 as a template Resolution: 2.16→43.2 Å / Cor.coef. Fo:Fc: 0.952 / Cor.coef. Fo:Fc free: 0.944 / SU R Cruickshank DPI: 0.185 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.183 / SU Rfree Blow DPI: 0.155 / SU Rfree Cruickshank DPI: 0.157
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 240.51 Å2 / Biso mean: 52.69 Å2 / Biso min: 30.86 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.25 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.16→43.2 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.16→2.22 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
