 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6rlt: Trypanosoma brucei Seryl-tRNA Synthetase in Complex with 5'-O-(N-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6rlt | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Trypanosoma brucei Seryl-tRNA Synthetase in Complex with 5'-O-(N-(L-seryl)-Sulfamoyl)uridine | ||||||||||||
 Components Components | Seryl-tRNA synthetase, putative | ||||||||||||
 Keywords Keywords | LIGASE / Coil-coil / Beta barrel / tRNA synthetase / Inhibitor / Complex | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationserine-tRNA ligase / serine-tRNA ligase activity / seryl-tRNA aminoacylation / ATP binding Similarity search - Function | ||||||||||||
| Biological species |  | ||||||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.84 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.84 Å | ||||||||||||
 Authors Authors | Pang, L. / De Graef, S. / Strelkov, S.V. / Weeks, S.D. | ||||||||||||
| Funding support |  Belgium, 3items Belgium, 3items
| ||||||||||||
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2020 Title: Structural Insights into the Binding of Natural Pyrimidine-Based Inhibitors of Class II Aminoacyl-tRNA Synthetases. Authors: Pang, L. / Nautiyal, M. / De Graef, S. / Gadakh, B. / Zorzini, V. / Economou, A. / Strelkov, S.V. / Van Aerschot, A. / Weeks, S.D. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6rlt.cif.gz | 382.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6rlt.ent.gz | 310.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6rlt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rl/6rltftp://data.pdbj.org/pub/pdb/validation_reports/rl/6rlt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6hdzC  6he1C  6he3C  6hhvC  6hhwC  6hhxC  6hhyC  6hhzC  6hi0C  6rluC  6rlvC  6s30C  6sjcC  3lsqS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 53923.203 Da / Num. of mol.: 2 / Fragment: Seryl-tRNA synthetase Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: MHOM/CI/86/DAL972 / Gene: TbgDal_XI8110 / Plasmid: pETRUK / Production host:  |
|---|
-Non-polymers , 5 types, 812 molecules 

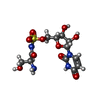
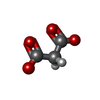

| #2: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Na#4: Chemical |  Mass: 410.357 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H18N4O10S / Feature type: SUBJECT OF INVESTIGATION Mass: 410.357 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H18N4O10S / Feature type: SUBJECT OF INVESTIGATION#5: Chemical |  Mass: 102.046 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H2O4 Mass: 102.046 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H2O4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 792 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.46 Å3/Da / Density % sol: 64.49 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7 Details: Holo protein at 10 mg/mL in 10 mM Tris pH 7, 100 mM NaCl, 5 mM DTT, 5% (v/v) glycerol was mixed with 2.4 M sodium malonate pH 7.0, 100 mM Bis-tris propane pH 7.0 in a 2:1 (v/v) ratio. ...Details: Holo protein at 10 mg/mL in 10 mM Tris pH 7, 100 mM NaCl, 5 mM DTT, 5% (v/v) glycerol was mixed with 2.4 M sodium malonate pH 7.0, 100 mM Bis-tris propane pH 7.0 in a 2:1 (v/v) ratio. Suitable crystals were soaked with 2 mM 5'-O-(N-(L-seryl)-Sulfamoyl)uridine in crystallization solution containing a 22% v/v glycerol. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.54179 Å | ||||||||||||||||||||||||||||||
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Nov 20, 2018 / Details: mirrors | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54179 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.84→130.43 Å / Num. obs: 127755 / % possible obs: 98.8 % / Redundancy: 10.7 % / Biso Wilson estimate: 16.12 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.082 / Rpim(I) all: 0.026 / Rrim(I) all: 0.086 / Net I/σ(I): 27.7 / Num. measured all: 1372322 / Scaling rejects: 229 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3LSQ Resolution: 1.84→21.2 Å / Cor.coef. Fo:Fc: 0.917 / Cor.coef. Fo:Fc free: 0.898 / SU R Cruickshank DPI: 0.115 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.121 / SU Rfree Blow DPI: 0.115 / SU Rfree Cruickshank DPI: 0.112
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 110.47 Å2 / Biso mean: 24.94 Å2 / Biso min: 6.28 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.23 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.84→21.2 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.84→1.86 Å / Rfactor Rfree error: 0 / Total num. of bins used: 50
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
