 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6hae: Crystal structure of [Fe]-hydrogenase (Hmd) from Methanococcus ae... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6hae | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of [Fe]-hydrogenase (Hmd) from Methanococcus aeolicus in complex with FeGP cofactor and methenyl-tetrahydromethanopterin (close form B) | ||||||
 Components Components | 5,10-methenyltetrahydromethanopterin hydrogenase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / [Fe]-hydrogenase / catalytic cycle / conformational rearrangement / Fe-guanylylpyridinol cofactor / methanogenesis / hydride-transfer / tetrahydromethanopterin / C1-metabolism | ||||||
| Function / homology |  Function and homology information Function and homology information5,10-methenyltetrahydromethanopterin hydrogenase / N5,N10-methenyltetrahydromethanopterin hydrogenase activity / methanogenesis, from carbon dioxide / pyrroline-5-carboxylate reductase activity / L-proline biosynthetic process / one-carbon metabolic process Similarity search - Function | ||||||
| Biological species | Methanococcus aeolicus | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å | ||||||
 Authors Authors | Huang, G. / Wagner, T. / Wodrich, M.D. / Ataka, K. / Bill, E. / Ermler, U. / Hu, X. / Shima, S. | ||||||
| Funding support |  Germany, 1items Germany, 1items
| ||||||
 Citation Citation | Journal: Nat Catal / Year: 2019 Title: The atomic-resolution crystal structure of activated [Fe]-hydrogenase Authors: Huang, G. / Wagner, T. / Wodrich, M.D. / Ataka, K. / Bill, E. / Ermler, U. / Hu, X. / Shima, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6hae.cif.gz | 169 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6hae.ent.gz | 129.4 KB | Display | PDB format |
| PDBx/mmJSON format | 6hae.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ha/6haeftp://data.pdbj.org/pub/pdb/validation_reports/ha/6hae | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6hacSC  6havC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AK
| #1: Protein | Mass: 36784.562 Da / Num. of mol.: 2 / Mutation: wild-type Source method: isolated from a genetically manipulated source Details: / Source: (gene. exp.)  Methanococcus aeolicus (strain ATCC BAA-1280 / DSM 17508 / OCM 812 / Nankai-3) (archaea) Methanococcus aeolicus (strain ATCC BAA-1280 / DSM 17508 / OCM 812 / Nankai-3) (archaea)Strain: ATCC BAA-1280 / DSM 17508 / OCM 812 / Nankai-3 / Tissue: / / Cell: / / Cell line: / / Gene: hmd, Maeo_1025 / Organ: / / Variant: / / Plasmid: pET-24b+ / Details (production host): / / Cell (production host): / / Cell line (production host): / / Organ (production host): / / Production host:  References: UniProt: A6UVT1, 5,10-methenyltetrahydromethanopterin hydrogenase |
|---|
-Non-polymers , 9 types, 622 molecules 
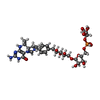

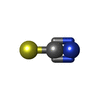


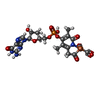
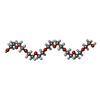

| #2: Chemical |  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical |  Mass: 787.685 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C31H44N6O16P Mass: 787.685 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C31H44N6O16P#4: Chemical |  Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#5: Chemical | ChemComp-SCN /  Mass: 58.082 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: CNS Mass: 58.082 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: CNS#6: Chemical |  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#7: Chemical | ChemComp-K /  Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K#8: Chemical |  Mass: 686.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H23FeN6O13PS Mass: 686.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H23FeN6O13PS#9: Chemical | ChemComp-2PE / |  Mass: 414.488 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H38O10 / Comment: precipitant*YM Mass: 414.488 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H38O10 / Comment: precipitant*YM#10: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 592 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.29 Å3/Da / Density % sol: 46.22 % / Description: Transparent plate-shape crystals |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 7 Details: Crystallization of [Fe]-hydrogenase-methenyl-H4MPT+ complex was performed in an anaerobic tent with gas phase 100%N2 at room temperature under dark condition. The reconstituted [Fe]- ...Details: Crystallization of [Fe]-hydrogenase-methenyl-H4MPT+ complex was performed in an anaerobic tent with gas phase 100%N2 at room temperature under dark condition. The reconstituted [Fe]-hydrogenase holoenzyme (50 mg/ml) was mixed with 10-mM methenyl-H4MPT+, both of which contained 10-mM MOPS/KOH pH 7.0. The final concentrations of [Fe]-hydrogenase and methenyl-H4MPT+ were 24 mg/ml and 3 mM, respectively. After incubating the mixture in this tent under dark condition for 5 min, the enzyme solution was centrifuged using MiniSpin-plus (Eppendorf) at 8000 rpm for 5 min by using centrifugal filters made of polyvinylidene fluoride (PVDF, Millipore). The crystallization solution contained 20 % w/v polyethylene glycol 3350, 200 mM sodium thiocyanate and 3% w/v 1,5-diaminopentane dihydrochloride with a ratio of protein mixture and crystallization reservoir of 0.7 ul / 0.7 ul drops (in 96 well-plates). PH range: / Temp details: The protein was crystallized at room temperature with about 3-4 degree of temperature variation |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 0.99992 Å / Beamline: X10SA / Wavelength: 0.99992 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jul 6, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.99992 Å / Relative weight: 1 |
| Reflection | Resolution: 1.85→44.56 Å / Num. obs: 56771 / % possible obs: 97.4 % / Redundancy: 6 % / Biso Wilson estimate: 26.94 Å2 / CC1/2: 0.997 / Rmerge(I) obs: 0.124 / Rpim(I) all: 0.055 / Rrim(I) all: 0.135 / Net I/σ(I): 8.6 |
| Reflection shell | Resolution: 1.85→1.95 Å / Redundancy: 4.1 % / Rmerge(I) obs: 1.133 / Mean I/σ(I) obs: 1.2 / Num. unique obs: 6994 / CC1/2: 0.323 / Rpim(I) all: 0.627 / Rrim(I) all: 1.301 / % possible all: 85 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6HAC Resolution: 1.85→44.56 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.959 / SU R Cruickshank DPI: 0.216 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.141 / SU Rfree Blow DPI: 0.121 / SU Rfree Cruickshank DPI: 0.116
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.83 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.19 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.85→44.56 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.85→1.9 Å / Total num. of bins used: 20
|
