 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6hac: Crystal structure of [Fe]-hydrogenase (Hmd) holoenzyme from Metha... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6hac | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of [Fe]-hydrogenase (Hmd) holoenzyme from Methanococcus aeolicus (open form) | ||||||
 Components Components | 5,10-methenyltetrahydromethanopterin hydrogenase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / [Fe]-hydrogenase / catalytic cycle / conformational rearrangement / Fe-guanylylpyridinol cofactor / methanogenesis / hydride-transfer / tetrahydromethanopterin / C1-metabolism | ||||||
| Function / homology |  Function and homology information Function and homology information5,10-methenyltetrahydromethanopterin hydrogenase / N5,N10-methenyltetrahydromethanopterin hydrogenase activity / methanogenesis, from carbon dioxide / pyrroline-5-carboxylate reductase activity / L-proline biosynthetic process / one-carbon metabolic process Similarity search - Function | ||||||
| Biological species | Methanococcus aeolicus | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Huang, G. / Wagner, T. / Wodrich, M.D. / Ataka, K. / Bill, E. / Ermler, U. / Hu, X. / Shima, S. | ||||||
| Funding support |  Germany, 1items Germany, 1items
| ||||||
 Citation Citation | Journal: Nat Catal / Year: 2019 Title: The atomic-resolution crystal structure of activated [Fe]-hydrogenase Authors: Huang, G. / Wagner, T. / Wodrich, M.D. / Ataka, K. / Bill, E. / Ermler, U. / Hu, X. / Shima, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6hac.cif.gz | 150.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6hac.ent.gz | 116.8 KB | Display | PDB format |
| PDBx/mmJSON format | 6hac.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ha/6hacftp://data.pdbj.org/pub/pdb/validation_reports/ha/6hac | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6haeC  6havC  4jjfS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 36784.562 Da / Num. of mol.: 1 / Mutation: wild-type Source method: isolated from a genetically manipulated source Details: / Source: (gene. exp.)  Methanococcus aeolicus (strain ATCC BAA-1280 / DSM 17508 / OCM 812 / Nankai-3) (archaea) Methanococcus aeolicus (strain ATCC BAA-1280 / DSM 17508 / OCM 812 / Nankai-3) (archaea)Tissue: / / Cell: / / Cell line: / / Gene: hmd, Maeo_1025 / Organ: / / Variant: / / Plasmid: pET-24b+ / Details (production host): / / Cell (production host): / / Cell line (production host): / / Organ (production host): / / Production host:  References: UniProt: A6UVT1, 5,10-methenyltetrahydromethanopterin hydrogenase |
|---|
-Non-polymers , 5 types, 112 molecules 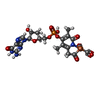




| #2: Chemical | ChemComp-FE9 /  Mass: 686.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H23FeN6O13PS Mass: 686.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H23FeN6O13PS | ||||
|---|---|---|---|---|---|
| #3: Chemical | ChemComp-1PE /  Mass: 238.278 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM Mass: 238.278 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM | ||||
| #4: Chemical |  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#5: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 107 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.74 Å3/Da / Density % sol: 67.17 % / Description: Transparent diamond shape |
|---|---|
| Crystal grow | Temperature: 281.15 K / Method: vapor diffusion, sitting drop / pH: 4 Details: The reconstituted [Fe]-hydrogenase holoenzyme from M. aeolicus was crystallized in the anaerobic tent with gas phase 95%N2/5%H2 using 96-well 2-drop MRC Crystallization Plates (Molecular ...Details: The reconstituted [Fe]-hydrogenase holoenzyme from M. aeolicus was crystallized in the anaerobic tent with gas phase 95%N2/5%H2 using 96-well 2-drop MRC Crystallization Plates (Molecular Dimensions, Suffolk, UK). For the initial screening, 0.7-ul of 24 mg/ml reconstituted [Fe]-hydrogenase was mixed with 0.7 ul of reservoir solution of crystallization kits under yellow light and incubated under the dark condition. The best diffracting crystal was obtained in one month in the crystallization reservoir solution containing 20% w/v polyethylene glycol 3350, 100-mM tri-sodium citrate pH 4.0 and 200-mM tri-sodium citrate. PH range: / Temp details: Temperature variation was +/- 1 during crystallization |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM30A / Wavelength: 0.9798 Å / Beamline: BM30A / Wavelength: 0.9798 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Sep 26, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9798 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→48.43 Å / Num. obs: 25078 / % possible obs: 100 % / Redundancy: 13.7 % / Biso Wilson estimate: 39.33 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.155 / Rpim(I) all: 0.043 / Rrim(I) all: 0.161 / Net I/σ(I): 17.9 |
| Reflection shell | Resolution: 2.3→2.42 Å / Redundancy: 14 % / Rmerge(I) obs: 1.023 / Mean I/σ(I) obs: 2.9 / Num. unique obs: 3607 / CC1/2: 0.602 / Rpim(I) all: 0.281 / Rrim(I) all: 1.061 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4JJF Resolution: 2.3→26.62 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.946 / SU R Cruickshank DPI: 0.351 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.177 / SU Rfree Blow DPI: 0.146 / SU Rfree Cruickshank DPI: 0.146
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 43.63 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.27 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.3→26.62 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.39 Å / Total num. of bins used: 13
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -2.1917 Å / Origin y: -41.5428 Å / Origin z: -22.9285 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: { A|* } |
