Entry Database : PDB / ID : 6h54Title CRYSTAL STRUCTURE OF BOVINE HSC70(AA1-554)E213A/D214A IN COMPLEX WITH INHIBITOR VER155008 Heat shock cognate 71 kDa protein Keywords / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Bos taurus (domestic cattle)Method / / / Resolution : 2.02 Å Authors Plank, C. / Zehe, M. / Grimm, C. / Sotriffer, C. Funding support Organization Grant number Country German Research Foundation (DFG) CRU 216
Journal : Acs Chem.Biol. / Year : 2024Title : Combined In-Solution Fragment Screening and Crystallographic Binding-Mode Analysis with a Two-Domain Hsp70 Construct.Authors : Zehe, M. / Kehrein, J. / Schollmayer, C. / Plank, C. / Kovacs, H. / Merino Asumendi, E. / Holzgrabe, U. / Grimm, C. / Sotriffer, C. History Deposition Jul 23, 2018 Deposition site / Processing site Revision 1.0 Aug 14, 2019 Provider / Type Revision 1.1 Aug 21, 2019 Group / Category / Item Revision 2.0 Jul 8, 2020 Group Advisory / Atomic model ... Advisory / Atomic model / Author supporting evidence / Data collection / Database references / Derived calculations / Non-polymer description / Other / Refinement description / Source and taxonomy / Structure summary Category atom_site / atom_site_anisotrop ... atom_site / atom_site_anisotrop / audit_author / cell / chem_comp / citation / citation_author / computing / diffrn / entity / entity_src_gen / pdbx_audit_support / pdbx_entity_instance_feature / pdbx_entity_nonpoly / pdbx_entry_details / pdbx_nonpoly_scheme / pdbx_refine_tls / pdbx_refine_tls_group / pdbx_struct_assembly_gen / pdbx_struct_assembly_prop / pdbx_struct_sheet_hbond / pdbx_validate_close_contact / pdbx_validate_symm_contact / pdbx_validate_torsion / refine / refine_hist / refine_ls_restr / refine_ls_shell / reflns / software / struct_asym / struct_conf / struct_mon_prot_cis / struct_site / struct_site_gen / symmetry Item _cell.angle_beta / _cell.volume ... _cell.angle_beta / _cell.volume / _chem_comp.formula / _chem_comp.formula_weight / _chem_comp.id / _chem_comp.mon_nstd_flag / _chem_comp.name / _chem_comp.pdbx_synonyms / _chem_comp.type / _citation.title / _diffrn.pdbx_serial_crystal_experiment / _entity_src_gen.gene_src_common_name / _pdbx_audit_support.funding_organization / _pdbx_struct_assembly_gen.asym_id_list / _pdbx_struct_assembly_prop.value / _pdbx_struct_sheet_hbond.range_1_auth_comp_id / _pdbx_struct_sheet_hbond.range_1_auth_seq_id / _pdbx_struct_sheet_hbond.range_1_label_comp_id / _pdbx_struct_sheet_hbond.range_1_label_seq_id / _pdbx_struct_sheet_hbond.range_2_auth_comp_id / _pdbx_struct_sheet_hbond.range_2_auth_seq_id / _pdbx_struct_sheet_hbond.range_2_label_comp_id / _pdbx_struct_sheet_hbond.range_2_label_seq_id / _refine.B_iso_mean / _refine.ls_R_factor_R_free / _refine.ls_R_factor_R_work / _refine.ls_R_factor_obs / _refine.ls_d_res_high / _refine.ls_number_reflns_R_work / _refine.overall_SU_ML / _refine.pdbx_method_to_determine_struct / _refine.pdbx_overall_phase_error / _refine.pdbx_starting_model / _refine.pdbx_stereochemistry_target_values / _refine.solvent_model_details / _refine_hist.d_res_high / _refine_hist.number_atoms_solvent / _refine_hist.number_atoms_total / _refine_hist.pdbx_number_atoms_ligand / _refine_hist.pdbx_number_atoms_protein / _refine_ls_restr.dev_ideal / _refine_ls_restr.number / _refine_ls_restr.type / _refine_ls_shell.R_factor_R_free / _refine_ls_shell.R_factor_R_work / _refine_ls_shell.d_res_high / _refine_ls_shell.d_res_low / _refine_ls_shell.percent_reflns_obs / _reflns.B_iso_Wilson_estimate / _struct_mon_prot_cis.pdbx_omega_angle / _symmetry.space_group_name_Hall Description / Provider / Type Revision 2.1 Jan 17, 2024 Group Advisory / Data collection ... Advisory / Data collection / Database references / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / pdbx_unobs_or_zero_occ_atoms Item / _database_2.pdbx_database_accessionRevision 2.2 Feb 14, 2024 Group / Category / citation_authorItem _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Germany, 1items
Germany, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads















 PDBj
PDBj
 Assembly
Assembly

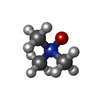
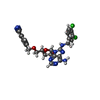



 Mass: 75.110 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H9NO
Mass: 75.110 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H9NO Mass: 556.401 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H23Cl2N7O4 / Feature type: SUBJECT OF INVESTIGATION
Mass: 556.401 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H23Cl2N7O4 / Feature type: SUBJECT OF INVESTIGATION Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM
Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Sample preparation
Sample preparation / Beamline: ID30B / Wavelength: 0.9763 Å
/ Beamline: ID30B / Wavelength: 0.9763 Å Processing
Processing