| 登録情報 | データベース: PDB / ID: 5z4m
|
|---|
| タイトル | Structure of TailorD343A with bound UTP and Mg |
|---|
 要素 要素 | Terminal uridylyltransferase Tailor |
|---|
 キーワード キーワード | TRANSFERASE / Terminal uridylyltransferase |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
negative regulation of pre-miRNA processing / positive regulation of miRNA catabolic process / RNA uridylyltransferase / RNA uridylyltransferase activity / regulatory ncRNA-mediated gene silencing / ATP binding / metal ion binding / cytoplasm類似検索 - 分子機能 Cid1 family poly A polymerase / : / Poly(A) RNA polymerase, mitochondrial-like, central palm domain / Nucleotidyltransferase superfamily類似検索 - ドメイン・相同性 URIDINE 5'-TRIPHOSPHATE / Terminal uridylyltransferase Tailor類似検索 - 構成要素 |
|---|
| 生物種 |   Drosophila melanogaster (キイロショウジョウバエ) Drosophila melanogaster (キイロショウジョウバエ) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.74 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.74 Å |
|---|
 データ登録者 データ登録者 | Cheng, L. / Li, F. / Jiang, Y. / Yu, H. / Xie, C. / Shi, Y. / Gong, Q. |
|---|
 引用 引用 | ジャーナル: Nucleic Acids Res. / 年: 2019
タイトル: Structural insights into a unique preference for 3' terminal guanine of mirtron in Drosophila TUTase tailor.
著者: Cheng, L. / Li, F. / Jiang, Y. / Yu, H. / Xie, C. / Shi, Y. / Gong, Q. |
|---|
| 履歴 | | 登録 | 2018年1月11日 | 登録サイト: PDBJ / 処理サイト: PDBJ |
|---|
| 改定 1.0 | 2018年10月31日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2018年12月19日 | Group: Data collection / Database references / カテゴリ: citation
Item: _citation.country / _citation.journal_abbrev ..._citation.country / _citation.journal_abbrev / _citation.journal_id_ASTM / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year |
|---|
| 改定 1.2 | 2019年1月23日 | Group: Data collection / Database references / カテゴリ: citation
Item: _citation.journal_volume / _citation.page_first ..._citation.journal_volume / _citation.page_first / _citation.page_last / _citation.year |
|---|
| 改定 1.3 | 2023年11月22日 | Group: Data collection / Database references / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード














 PDBj
PDBj 集合体
集合体

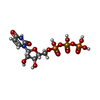
 分子量: 484.141 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C9H15N2O15P3 / コメント: UTP*YM
分子量: 484.141 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C9H15N2O15P3 / コメント: UTP*YM
 分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg
分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg 分子量: 18.015 Da / 分子数: 177 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 177 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: BL19U1 / 波長: 0.9785 Å
/ ビームライン: BL19U1 / 波長: 0.9785 Å 解析
解析