Entry Database : PDB / ID : 5z0bTitle Crystal structure of plasma-derived human serum albumin Serum albumin Keywords / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 2.17 Å Authors Park, J. / Kim, M.-S. / Shin, D.H. Funding support Organization Grant number Country National Research Foundation (NRF, Korea) 2015R1D1A1A01058942
Journal : To Be Published Title : The crystal structure of plasma-derived human serum albuminAuthors : Park, J. / Kim, M.S. / Shin, D.H. History Deposition Dec 19, 2017 Deposition site / Processing site Revision 1.0 Dec 19, 2018 Provider / Type Revision 2.0 Jun 24, 2020 Group Advisory / Atomic model ... Advisory / Atomic model / Author supporting evidence / Data collection / Derived calculations / Non-polymer description / Other / Refinement description / Structure summary Category atom_site / atom_site_anisotrop ... atom_site / atom_site_anisotrop / cell / chem_comp / computing / diffrn / entity / pdbx_audit_support / pdbx_entity_instance_feature / pdbx_entity_nonpoly / pdbx_entry_details / pdbx_nonpoly_scheme / pdbx_refine_tls / pdbx_refine_tls_group / pdbx_struct_assembly_gen / pdbx_struct_assembly_prop / pdbx_validate_close_contact / pdbx_validate_symm_contact / pdbx_validate_torsion / refine / refine_hist / refine_ls_restr / refine_ls_shell / reflns / software / struct_asym / struct_conf / struct_conn / struct_mon_prot_cis / struct_site / struct_site_gen / symmetry Item _cell.angle_beta / _cell.volume ... _cell.angle_beta / _cell.volume / _chem_comp.formula / _chem_comp.formula_weight / _chem_comp.id / _chem_comp.mon_nstd_flag / _chem_comp.name / _chem_comp.pdbx_synonyms / _chem_comp.type / _diffrn.pdbx_serial_crystal_experiment / _pdbx_audit_support.funding_organization / _pdbx_struct_assembly_gen.asym_id_list / _pdbx_struct_assembly_prop.value / _pdbx_validate_symm_contact.auth_asym_id_1 / _pdbx_validate_symm_contact.auth_asym_id_2 / _pdbx_validate_symm_contact.auth_atom_id_1 / _pdbx_validate_symm_contact.auth_atom_id_2 / _pdbx_validate_symm_contact.auth_comp_id_1 / _pdbx_validate_symm_contact.auth_comp_id_2 / _pdbx_validate_symm_contact.auth_seq_id_1 / _pdbx_validate_symm_contact.auth_seq_id_2 / _pdbx_validate_symm_contact.dist / _pdbx_validate_symm_contact.site_symmetry_2 / _refine.B_iso_mean / _refine.ls_R_factor_R_free / _refine.ls_R_factor_R_work / _refine.ls_R_factor_obs / _refine.ls_d_res_high / _refine.ls_number_reflns_R_work / _refine.ls_number_reflns_all / _refine.overall_SU_ML / _refine.pdbx_overall_phase_error / _refine.pdbx_stereochemistry_target_values / _refine.solvent_model_details / _refine_hist.d_res_high / _refine_hist.number_atoms_solvent / _refine_hist.number_atoms_total / _refine_hist.pdbx_number_atoms_ligand / _refine_ls_restr.dev_ideal / _refine_ls_restr.number / _refine_ls_restr.type / _refine_ls_shell.R_factor_R_free / _refine_ls_shell.R_factor_R_work / _refine_ls_shell.d_res_high / _refine_ls_shell.d_res_low / _refine_ls_shell.number_reflns_R_free / _refine_ls_shell.number_reflns_R_work / _refine_ls_shell.percent_reflns_obs / _reflns.B_iso_Wilson_estimate / _software.version / _struct_mon_prot_cis.pdbx_omega_angle / _symmetry.space_group_name_Hall Description Details Provider / Type Revision 2.1 Nov 22, 2023 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accessionRevision 2.2 Oct 23, 2024 Group / Category / pdbx_modification_feature / Item
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Korea, Republic Of, 1items
Korea, Republic Of, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj

















 Assembly
Assembly

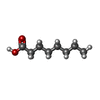

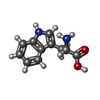






 Mass: 280.445 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C18H32O2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 280.445 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C18H32O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 144.211 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C8H16O2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 144.211 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C8H16O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 256.424 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C16H32O2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 256.424 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C16H32O2 / Feature type: SUBJECT OF INVESTIGATION Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H12N2O2 / Feature type: SUBJECT OF INVESTIGATION
Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H12N2O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 96.063 Da / Num. of mol.: 19 / Source method: isolated from a natural source / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 19 / Source method: isolated from a natural source / Formula: SO4 Mass: 106.120 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 150.173 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H14O4
Mass: 150.173 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 194.226 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM
Mass: 194.226 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 238.278 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM
Mass: 238.278 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H22O6 / Comment: precipitant*YM Sample preparation
Sample preparation / Beamline: MX1 / Wavelength: 0.9536 Å
/ Beamline: MX1 / Wavelength: 0.9536 Å Processing
Processing