Journal: Sci Rep / Year: 2017 Title: Visualisation of a flexible modular structure of the ER folding-sensor enzyme UGGT. Authors: Tadashi Satoh / Chihong Song / Tong Zhu / Takayasu Toshimori / Kazuyoshi Murata / Yugo Hayashi / Hironari Kamikubo / Takayuki Uchihashi / Koichi Kato / Abstract: In the endoplasmic reticulum (ER), a protein quality control system facilitates the efficient folding of newly synthesised proteins. In this system, a series of N-linked glycan intermediates ...In the endoplasmic reticulum (ER), a protein quality control system facilitates the efficient folding of newly synthesised proteins. In this system, a series of N-linked glycan intermediates displayed on the protein surface serve as quality tags. The ER folding-sensor enzyme UDP-glucose:glycoprotein glucosyltransferase (UGGT) acts as a gatekeeper in the ER quality control system by specifically catalysing monoglucosylation onto incompletely folded glycoproteins, thereby enabling them to interact with lectin-chaperone complexes. Here we characterise the dynamic structure of this enzyme. Our crystallographic data demonstrate that the sensor region is composed of four thioredoxin-like domains followed by a β-rich domain, which are arranged into a C-shaped structure with a large central cavity, while the C-terminal catalytic domain undergoes a ligand-dependent conformational alteration. Furthermore, small-angle X-ray scattering, cryo-electron microscopy and high-speed atomic force microscopy have demonstrated that UGGT has a flexible modular structure in which the smaller catalytic domain is tethered to the larger folding-sensor region with variable spatial arrangements. These findings provide structural insights into the working mechanism whereby UGGT operates as a folding-sensor against a variety of glycoprotein substrates through its flexible modular structure possessing extended hydrophobic surfaces for the recognition of unfolded substrates.
Resolution: 1.35→20 Å / Cor.coef. Fo:Fc: 0.965 / Cor.coef. Fo:Fc free: 0.948 / SU B: 2.155 / SU ML: 0.039 / Cross valid method: THROUGHOUT / ESU R: 0.061 / ESU R Free: 0.059 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.20759
3112
5 %
RANDOM
Rwork
0.1686
-
-
-
obs
0.17054
59135
99.08 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Thermomyces dupontii (fungus)
Thermomyces dupontii (fungus) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Japan, 3items
Japan, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj


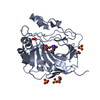
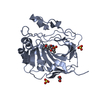
 Assembly
Assembly

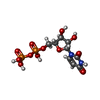
 Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM
Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM
 Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM
Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM
 Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 18.015 Da / Num. of mol.: 220 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 220 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing