Entry Database : PDB / ID : 5tmaTitle Zymomonas mobilis pyruvate decarboxylase mutant PDC-2.3 Pyruvate decarboxylase Keywords / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Zymomonas mobilis (bacteria)Method / / Resolution : 1.67 Å Authors Alahuhta, P.M. / Lunin, V.V. Journal : Biotechnol Biofuels / Year : 2018Title : An iterative computational design approach to increase the thermal endurance of a mesophilic enzyme.Authors : Sammond, D.W. / Kastelowitz, N. / Donohoe, B.S. / Alahuhta, M. / Lunin, V.V. / Chung, D. / Sarai, N.S. / Yin, H. / Mittal, A. / Himmel, M.E. / Guss, A.M. / Bomble, Y.J. History Deposition Oct 12, 2016 Deposition site / Processing site Revision 1.0 Oct 18, 2017 Provider / Type Revision 1.1 Jun 5, 2019 Group / Database references / Derived calculationsCategory / citation_author / pdbx_struct_special_symmetryItem _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year Revision 1.2 Oct 4, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / struct_conn Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Zymomonas mobilis (bacteria)
Zymomonas mobilis (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj



 Assembly
Assembly

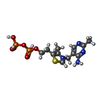




 Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S
Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 96.063 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: SO4 Mass: 62.068 Da / Num. of mol.: 18 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 18 / Source method: obtained synthetically / Formula: C2H6O2 Sample preparation
Sample preparation Processing
Processing