 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5o4m: Fresh crystals of HcgC from Methanococcus maripaludis cocrystalli... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5o4m | ||||||
|---|---|---|---|---|---|---|---|
| Title | Fresh crystals of HcgC from Methanococcus maripaludis cocrystallized with SAH and pyridinol | ||||||
 Components Components | HcgC | ||||||
 Keywords Keywords | TRANSFERASE / methyltransferases / biosynthesis / protein structures / enzyme catalysis / mutagenesis / [Fe]-hydrogenase / pyridinol / Hmd | ||||||
| Function / homology | FeGP cofactor biosynthesis protein, methyltransferase HcgC / FeGP cofactor biosynthesis protein, methyltransferase HcgC / 6-carboxy methyl-4-hydroxy-2-pyridinol / S-ADENOSYL-L-HOMOCYSTEINE / Uncharacterized protein Function and homology information Function and homology information | ||||||
| Biological species |  Methanococcus maripaludis S2 (archaea) Methanococcus maripaludis S2 (archaea) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Wagner, T. / Bai, L. / Xu, T. / Hu, X. / Ermler, U. / Shima, S. | ||||||
 Citation Citation | Journal: Angew. Chem. Int. Ed. Engl. / Year: 2017 Title: A Water-Bridged H-Bonding Network Contributes to the Catalysis of the SAM-Dependent C-Methyltransferase HcgC. Authors: Bai, L. / Wagner, T. / Xu, T. / Hu, X. / Ermler, U. / Shima, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5o4m.cif.gz | 446.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5o4m.ent.gz | 366.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5o4m.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/o4/5o4mftp://data.pdbj.org/pub/pdb/validation_reports/o4/5o4m | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5o4hSC  5o4jC  5o4nC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | Mass: 30942.623 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Details: / / Source: (gene. exp.) Methanococcus maripaludis S2 (archaea) / Tissue: / / Cell: / / Cell line: / / Gene: MMP1498 / Organ: / / Production host:  |
|---|
-Non-polymers , 5 types, 538 molecules 
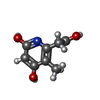



| #2: Chemical | ChemComp-SAH /  Mass: 384.411 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C14H20N6O5S Mass: 384.411 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C14H20N6O5S#3: Chemical | ChemComp-9KH /  Mass: 183.161 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H9NO4 Mass: 183.161 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H9NO4#4: Chemical | ChemComp-MPD / (  Mass: 118.174 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#5: Chemical |  Mass: 78.133 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 522 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.29 Å3/Da / Density % sol: 46.3 % / Description: Thick transparent hexagonal brick |
|---|---|
| Crystal grow | Temperature: 281 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: The drop consists of 1 ul of enzyme solution containing ~5 mg/ml HcgC, 2 mM pyridinol (dissolved in 100% DMSO) and 2 mM SAH mixed with 1 ul of the reservoir solution : 100 mM HEPES/NaOH pH 7. ...Details: The drop consists of 1 ul of enzyme solution containing ~5 mg/ml HcgC, 2 mM pyridinol (dissolved in 100% DMSO) and 2 mM SAH mixed with 1 ul of the reservoir solution : 100 mM HEPES/NaOH pH 7.5, 0.1 M NaCl, and 30% 2-Methyl-2,4-pentanediol. After only 4 days the crystals were immediately fished and frozen in liquid nitrogen for synchrotron radiation. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 1.00001 Å / Beamline: X10SA / Wavelength: 1.00001 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Feb 17, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.00001 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→47.11 Å / Num. obs: 63739 / % possible obs: 97.9 % / Redundancy: 2.6 % / Biso Wilson estimate: 40.63 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.054 / Rpim(I) all: 0.039 / Net I/σ(I): 11.5 |
| Reflection shell | Resolution: 2.1→2.21 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.544 / Mean I/σ(I) obs: 1.7 / Num. unique obs: 9372 / CC1/2: 0.804 / Rpim(I) all: 0.412 / % possible all: 98.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5O4H Resolution: 2.1→46.68 Å / Cor.coef. Fo:Fc: 0.9475 / Cor.coef. Fo:Fc free: 0.9206 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.237 / SU Rfree Blow DPI: 0.181
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 56.2 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.368 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.1→46.68 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.15 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
