 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5mny | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Neutron structure of cationic trypsin in complex with aniline | |||||||||
 Components Components | Cationic trypsin | |||||||||
 Keywords Keywords | HYDROLASE / hydrogen bonding / protonation / protein-ligand interaction | |||||||||
| Function / homology |  Function and homology information Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | NEUTRON DIFFRACTION / NUCLEAR REACTOR /  MOLECULAR REPLACEMENT / Resolution: 1.43 Å MOLECULAR REPLACEMENT / Resolution: 1.43 Å | |||||||||
 Authors Authors | Schiebel, J. / Schrader, T.E. / Ostermann, A. / Heine, A. / Klebe, G. | |||||||||
| Funding support | 1items
| |||||||||
 Citation Citation | Journal: Angew. Chem. Int. Ed. Engl. / Year: 2017 Title: Charges Shift Protonation: Neutron Diffraction Reveals that Aniline and 2-Aminopyridine Become Protonated Upon Binding to Trypsin. Authors: Schiebel, J. / Gaspari, R. / Sandner, A. / Ngo, K. / Gerber, H.D. / Cavalli, A. / Ostermann, A. / Heine, A. / Klebe, G. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5mny.cif.gz | 99.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5mny.ent.gz | 75.6 KB | Display | PDB format |
| PDBx/mmJSON format | 5mny.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mn/5mnyftp://data.pdbj.org/pub/pdb/validation_reports/mn/5mny | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5mn1C  5mnaC  5mnbC  5mncC  5mnxC  5monC  5mooC  4i8hS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23324.287 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #3: Chemical | ChemComp-WOT / 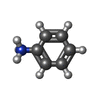  Mass: 94.134 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8N Mass: 94.134 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8N |
| #4: Chemical | ChemComp-DOD / 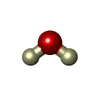  Mass: 18.015 Da / Num. of mol.: 129 / Source method: isolated from a natural source / Formula: D2O Mass: 18.015 Da / Num. of mol.: 129 / Source method: isolated from a natural source / Formula: D2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: NEUTRON DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 0.2 M ammonium sulfate, 0.1 M Hepes pH 7.5, 16.5% (w/v) PEG 8000 |
|---|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: NUCLEAR REACTOR / Site: FRM II  / Beamline: BIODIFF / Wavelength: 2.667 Å / Beamline: BIODIFF / Wavelength: 2.667 Å |
| Detector | Type: MAATEL BIODIFF / Detector: IMAGE PLATE / Date: Jul 22, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: neutron |
| Radiation wavelength | Wavelength: 2.667 Å / Relative weight: 1 |
| Reflection | Resolution: 1.43→50 Å / Num. obs: 38342 / % possible obs: 93.8 % / Redundancy: 2.5 % / Rmerge(I) obs: 0.098 / Net I/σ(I): 7.253 |
| Reflection shell | Resolution: 1.43→1.45 Å / Redundancy: 1.6 % / Rmerge(I) obs: 0.387 / Mean I/σ(I) obs: 1.858 / % possible all: 82.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4I8H Resolution: 1.43→22.09 Å / SU ML: 0.15 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 16.86
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
