 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5cfs: Crystal Structure of ANT(2")-Ia in complex with AMPCPP and tobramycin -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5cfs | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of ANT(2")-Ia in complex with AMPCPP and tobramycin | ||||||
 Components Components | AAD(2''),Gentamicin 2''-nucleotidyltransferase,Gentamicin resistance protein | ||||||
 Keywords Keywords | transferase/antibiotic / antibiotic resistance / nucleotidyltransferase / AMPCPP / tobramycin / Rossmann fold / transferase-antibiotic complex | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.7 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.7 Å | ||||||
 Authors Authors | Rodionov, D. / Bassenden, A.V. / Berghuis, A.M. | ||||||
| Funding support |  Canada, 1items Canada, 1items
| ||||||
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2016 Title: Structural Analysis of the Tobramycin and Gentamicin Clinical Resistome Reveals Limitations for Next-generation Aminoglycoside Design. Authors: Bassenden, A.V. / Rodionov, D. / Shi, K. / Berghuis, A.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5cfs.cif.gz | 97.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5cfs.ent.gz | 70.5 KB | Display | PDB format |
| PDBx/mmJSON format | 5cfs.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cf/5cfsftp://data.pdbj.org/pub/pdb/validation_reports/cf/5cfs | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5cftC  4xjeS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 20964.518 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria) / Gene: aadB, TNCP6 / Plasmid: pET-22b(+) / Production host: |
|---|
-Non-polymers , 6 types, 181 molecules 

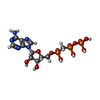
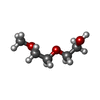
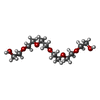

| #2: Chemical |  Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn#3: Chemical | ChemComp-TOY / |  Mass: 467.514 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H37N5O9 / Comment: antibiotic*YM Mass: 467.514 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H37N5O9 / Comment: antibiotic*YM#4: Chemical | ChemComp-APC / |  Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM#5: Chemical | ChemComp-PG0 / |  Mass: 120.147 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H12O3 / Comment: precipitant*YM Mass: 120.147 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H12O3 / Comment: precipitant*YM#6: Chemical | ChemComp-P6G / |  Mass: 282.331 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM Mass: 282.331 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 175 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.13 Å3/Da / Density % sol: 42.25 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 6.3 Details: 100 mM MES pH 6.3, 28% PEG 200, 5% PEG 3000, 1mM MnCl2, 10 mM AMPCPP, 10mM tobramycin |
-Data collection
| Diffraction | Mean temperature: 93 K | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.54178 Å | ||||||||||||||||||||||||
| Detector | Type: RIGAKU SATURN 944+ / Detector: CCD / Date: Feb 27, 2015 / Details: 2x2 binning | ||||||||||||||||||||||||
| Radiation | Monochromator: Rigaku VariMax-HF focusing mirror / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54178 Å / Relative weight: 1 | ||||||||||||||||||||||||
| Reflection | Resolution: 1.7→37.1 Å / Num. obs: 19150 / % possible obs: 99.7 % / Redundancy: 25.6 % / Biso Wilson estimate: 17.82 Å2 / Rmerge(I) obs: 0.055 / Rpim(I) all: 0.011 / Net I/σ(I): 43.1 / Num. measured all: 489780 | ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4XJE Resolution: 1.7→37.096 Å / SU ML: 0.15 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 18.98 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 56.59 Å2 / Biso mean: 22.9559 Å2 / Biso min: 10.03 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.7→37.096 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 7
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
