National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
GM071747
United States
Citation
Journal: Cell / Year: 2015 Title: An Atypical AAA+ ATPase Assembly Controls Efficient Transposition through DNA Remodeling and Transposase Recruitment. Authors: Ernesto Arias-Palomo / James M Berger / Abstract: Transposons are ubiquitous genetic elements that drive genome rearrangements, evolution, and the spread of infectious disease and drug-resistance. Many transposons, such as Mu, Tn7, and IS21, require ...Transposons are ubiquitous genetic elements that drive genome rearrangements, evolution, and the spread of infectious disease and drug-resistance. Many transposons, such as Mu, Tn7, and IS21, require regulatory AAA+ ATPases for function. We use X-ray crystallography and cryo-electron microscopy to show that the ATPase subunit of IS21, IstB, assembles into a clamshell-shaped decamer that sandwiches DNA between two helical pentamers of ATP-associated AAA+ domains, sharply bending the duplex into a 180° U-turn. Biochemical studies corroborate key features of the structure and further show that the IS21 transposase, IstA, recognizes the IstB•DNA complex and promotes its disassembly by stimulating ATP hydrolysis. Collectively, these studies reveal a distinct manner of higher-order assembly and client engagement by a AAA+ ATPase and suggest a mechanistic model where IstB binding and subsequent DNA bending primes a selected insertion site for efficient transposition.
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Geobacillus stearothermophilus (bacteria)
Geobacillus stearothermophilus (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj


 Assembly
Assembly




 Mass: 427.201 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM
Mass: 427.201 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM
 Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg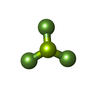
 Mass: 66.007 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: BeF3
Mass: 66.007 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: BeF3 Mass: 18.015 Da / Num. of mol.: 530 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 530 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing