 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5aqt: Fragment-based screening of HSP70 sheds light on the functional r... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5aqt | ||||||
|---|---|---|---|---|---|---|---|
| Title | Fragment-based screening of HSP70 sheds light on the functional role of ATP-binding site residues | ||||||
 Components Components |
| ||||||
 Keywords Keywords | CHAPERONE / HEAT SHOCK PROTEIN / HSP70 / HSP72 / HSC70 / ATPASE / BAG1 / FRAGMENT | ||||||
| Function / homology |  Function and homology information Function and homology informationlumenal side of lysosomal membrane / regulation of protein import / negative regulation of supramolecular fiber organization / chaperone-mediated autophagy translocation complex disassembly / clathrin-sculpted gamma-aminobutyric acid transport vesicle membrane / positive regulation of ferroptosis / Lipophagy / GABA synthesis, release, reuptake and degradation / protein targeting to lysosome involved in chaperone-mediated autophagy / Respiratory syncytial virus genome transcription ...lumenal side of lysosomal membrane / regulation of protein import / negative regulation of supramolecular fiber organization / chaperone-mediated autophagy translocation complex disassembly / clathrin-sculpted gamma-aminobutyric acid transport vesicle membrane / positive regulation of ferroptosis / Lipophagy / GABA synthesis, release, reuptake and degradation / protein targeting to lysosome involved in chaperone-mediated autophagy / Respiratory syncytial virus genome transcription / adenyl-nucleotide exchange factor activity / clathrin coat disassembly / C3HC4-type RING finger domain binding / positive regulation of smooth muscle cell apoptotic process / CHL1 interactions / regulation of protein complex stability / negative regulation of NLRP3 inflammasome complex assembly / ATP-dependent protein disaggregase activity / membrane organization / cellular response to steroid hormone stimulus / Lysosome Vesicle Biogenesis / chaperone-mediated autophagy / Golgi Associated Vesicle Biogenesis / non-chaperonin molecular chaperone ATPase / Prp19 complex / HSF1-dependent transactivation / Regulation of HSF1-mediated heat shock response / protein folding chaperone complex / response to unfolded protein / regulation of protein-containing complex assembly / Attenuation phase / ATP metabolic process / Protein methylation / heat shock protein binding / mRNA Splicing - Major Pathway / protein folding chaperone / lysosomal lumen / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / cellular response to starvation / spliceosomal complex / ATP-dependent protein folding chaperone / regulation of protein stability / AUF1 (hnRNP D0) binds and destabilizes mRNA / mRNA splicing, via spliceosome / Late endosomal microautophagy / PKR-mediated signaling / G protein-coupled receptor binding / Chaperone Mediated Autophagy / : / melanosome / MHC class II protein complex binding / Clathrin-mediated endocytosis / protein refolding / protein-folding chaperone binding / protein folding / Interleukin-4 and Interleukin-13 signaling / secretory granule lumen / blood microparticle / ficolin-1-rich granule lumen / protein-macromolecule adaptor activity / cell surface receptor signaling pathway / protein stabilization / positive regulation of cell migration / cadherin binding / ribonucleoprotein complex / receptor ligand activity / lysosomal membrane / focal adhesion / negative regulation of DNA-templated transcription / apoptotic process / ubiquitin protein ligase binding / Neutrophil degranulation / negative regulation of apoptotic process / nucleolus / enzyme binding / ATP hydrolysis activity / : / RNA binding / extracellular exosome / extracellular region / nucleoplasm / ATP binding / membrane / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Jones, A.M. / Westwood, I.M. / Osborne, J.D. / Matthews, T.P. / Cheeseman, M.D. / Rowlands, M.G. / Jeganathan, F. / Burke, R. / Lee, D. / Kadi, N. ...Jones, A.M. / Westwood, I.M. / Osborne, J.D. / Matthews, T.P. / Cheeseman, M.D. / Rowlands, M.G. / Jeganathan, F. / Burke, R. / Lee, D. / Kadi, N. / Liu, M. / Richards, M. / McAndrew, C. / Yahya, N. / Dobson, S.E. / Jones, K. / Workman, P. / Collins, I. / van Montfort, R.L.M. | ||||||
 Citation Citation | Journal: Sci Rep / Year: 2016 Title: A fragment-based approach applied to a highly flexible target: Insights and challenges towards the inhibition of HSP70 isoforms. Authors: Jones, A.M. / Westwood, I.M. / Osborne, J.D. / Matthews, T.P. / Cheeseman, M.D. / Rowlands, M.G. / Jeganathan, F. / Burke, R. / Lee, D. / Kadi, N. / Liu, M. / Richards, M. / McAndrew, C. / ...Authors: Jones, A.M. / Westwood, I.M. / Osborne, J.D. / Matthews, T.P. / Cheeseman, M.D. / Rowlands, M.G. / Jeganathan, F. / Burke, R. / Lee, D. / Kadi, N. / Liu, M. / Richards, M. / McAndrew, C. / Yahya, N. / Dobson, S.E. / Jones, K. / Workman, P. / Collins, I. / van Montfort, R.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5aqt.cif.gz | 210.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5aqt.ent.gz | 168.7 KB | Display | PDB format |
| PDBx/mmJSON format | 5aqt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/aq/5aqtftp://data.pdbj.org/pub/pdb/validation_reports/aq/5aqt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5aqfC  5aqgC  5aqhC  5aqiC  5aqjC  5aqkC  5aqlC  5aqmC  5aqnC  5aqoC  5aqpC  5aqqC  5aqrC  5aqsC  5aquC  5aqvC  5aqwC  5aqxC  5aqyC  1hx1S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj
















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 42406.980 Da / Num. of mol.: 1 / Fragment: NUCLEOTIDE BINDING DOMAIN, UNP RESIDUES 1-381 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PGEX-6P-1 / Production host:  |
|---|---|
| #2: Protein | Mass: 13511.571 Da / Num. of mol.: 1 / Fragment: UNP RESIDUES 222-334 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PGEX-6P-1 / Production host: |
-Non-polymers , 5 types, 220 molecules 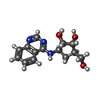




| #3: Chemical | ChemComp-5P7 / ( Mass: 275.303 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H17N3O3 Mass: 275.303 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H17N3O3 | ||||
|---|---|---|---|---|---|
| #4: Chemical | ChemComp-TRS /  Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM | ||||
| #5: Chemical |  Mass: 78.133 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#6: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C3H8O3#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 210 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.45 Å3/Da / Density % sol: 49.83 % / Description: NONE |
|---|---|
| Crystal grow | pH: 8.5 Details: 16-26% (W/V) PEG3350, 0.1 M K-NA TARTRATE, 0.1 M TRIS.HCL PH 8.5 AND 25% (V/V) GLYCEROL |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04-1 / Wavelength: 0.92 / Beamline: I04-1 / Wavelength: 0.92 |
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: Sep 29, 2012 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→58.26 Å / Num. obs: 36309 / % possible obs: 83.9 % / Observed criterion σ(I): 0 / Redundancy: 3 % / Biso Wilson estimate: 28.21 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 9.3 |
| Reflection shell | Resolution: 1.9→1.94 Å / Redundancy: 3.2 % / Rmerge(I) obs: 0.61 / Mean I/σ(I) obs: 1.7 / % possible all: 70.6 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1HX1 Resolution: 1.9→58.26 Å / Cor.coef. Fo:Fc: 0.945 / Cor.coef. Fo:Fc free: 0.917 / SU R Cruickshank DPI: 0.163 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.164 / SU Rfree Blow DPI: 0.149 / SU Rfree Cruickshank DPI: 0.149
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 40.54 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.236 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→58.26 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.96 Å / Total num. of bins used: 18
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
